文章概要
文章的题目,是《Genome-wide functional screen of 3‘UTR variants uncovers causal variants for human disease and evolution》
这篇文章,发表在Cell杂志2021年9月刊上
文章的题目翻译成中文,意思是《3'UTR 变异的全基因组功能筛选揭示了人类疾病和进化的因果变异》
文章的通讯作者,名字叫James R. Xue,是美国 Broad 研究所的研究人员。
研究背景
实验结果分析
实验内容的第一部分,MPRAu 可重现地表征数千个 3』UTR 元素的功能

MPRAu,是 massive parallel reporter assay for 3』UTR 的首字母缩写,翻译成中文,意思是「对 3』UTR 的大规模平行报告检测」。
接下来,会多次提到这个 MPRAu 方法,我会把它称为 M 方法,同样,提到 MPRAu 值,我们把它简称为「M 值」,以方便讲解
实验的第一步,是选取大量的目标的 3』UTR 区域的 100BP 长的片段,在芯片上进行大规模平行合成,在合成中既要合成参考型的基因序列,又要合成变异型的基因序列。
并且,在每个序列头上,还要加上一个特定的 Barcode 序列,有了这个 barcode 序列,在后面的实验中,可以通过跟踪 barcode 序列,来方便地回朔到原来的 3』UTR 序列。
在这其中,为了减少 barcode 序列造成的影响,作者对一对等位基因,用好几个随机的 barcode,以减小 barcode 序列造成的影响。
第二步,把合成好的 3』UTR 序列,连同 barcode 序列,组装进质粒。
这个质粒上已经事先连上了 GFP 蛋白的基因,也就是会发出荧光的报告基因。
第三步,把质粒转染进培养的细胞中去,在本项研究中,是转染了 6 种细胞系。
第四步,从培养的细胞中分离出 mRNA,并且进行扩增。
接下来,对 mRNA,和 DNA 进行高通量测序,然后,分析每种质粒,计算它的 mRNA 的产量,和它的质粒的数量的比值。
然后,比较参考虑序列的比值,和变异序列的比值,看这个差值的大小。
作者把这个差值,称作 allelic skew,翻成中文,是「等位基因倾斜」的意思。
作者一共是挑了 12,173 个 3』UTR 的变异,来做这项分析。被挑选出来的变异,是在 GWAS 分析中,被发现与疾病的发病有关联的 3』UTR 的 SNP 或者 indel
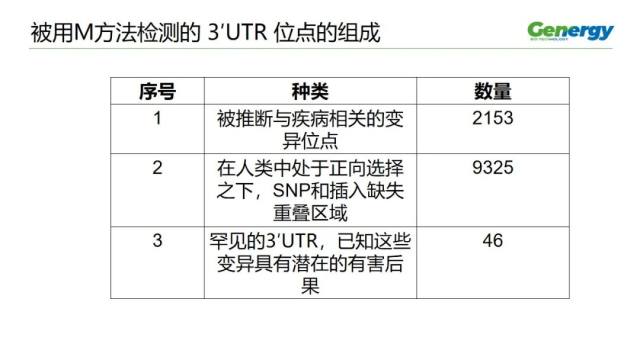
作者纳入 M 方法检测的基因位点,一共是 12173 个。
这其中的组成,大体分成三组,
1、被推断与疾病相关的位点,有 2153 个
2、在人类中处于正向选择之下的,SNP 和插入缺失重叠区域,有 9325 个,也就是更有利于人类生存的 SNP 和 indel 重叠区域
3、罕见的 3′UTR,已知这些变异具有潜在的有害后果,46 个

这是 RNA 计数与质粒 DNA 计数的关系,纵轴是 RNA 的计数,横轴是 DNA 的计数。
我们可以看到有较多的点落在了主线的下方,也就是说,这些 RNA 的表达,是有所减少的。
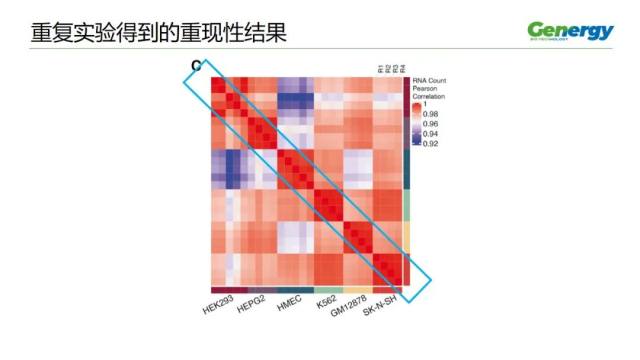
这是 6 种细胞,做重复实验,对各组重复实验进行两两比较,得到的热图。
横轴和纵轴上,分别标了 6 种色块,每一个色块,就是一种细胞。
我们可以看到,从左上到右下的这条斜的轴,是深红色的,
也就是说,同一种细胞的重复实验组,它们的内部的一致性很高,也就是重现性很好。
平均的皮尔森相关性系数达到 0.99
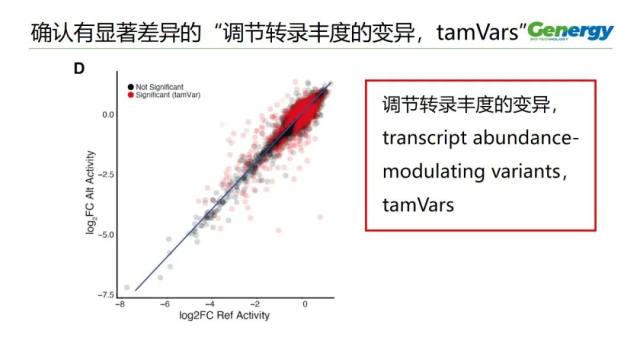
这张图,显示的是有显著差异的「调节转录丰度的变异」,它的英文原文是 transcript abundance-modulating variants,缩写是 tamVars
它显示的是变异的序列,和参考的序列,所导致的目标基因的 mRNA 表达量的差异。
图中横轴是参考序列,所产生的基因表达量;纵轴是变异的序列,所产生的基因表达量。
图中,每一个点,就是一对等位基因序列,它们的两种基因序列所产生的表达量,在 2 维座标中的交叉点。
图中,黑色的点是差异不显著的点,红色的点是差异显著的点。
作者发现了在所有 6 种细胞中都有的 2368 个 tamVars,
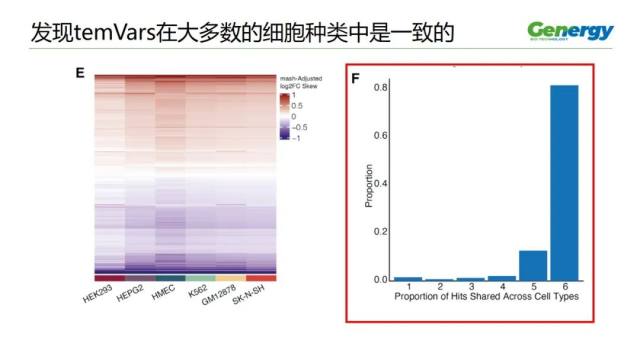
作者接着做了 temVars 在大多数的细胞种类中的一致性,
结果发现,81.2% 的 temVars 在 6 种细胞中共有,相比之下,1.6% 的 temVars 只在一种细胞中存在。
右边的 F 图,展示了 temVars 在几种细胞中共有的分布情况。
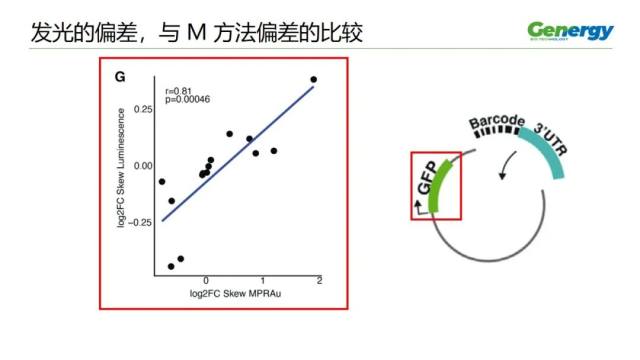
接下来,作者做了发光的偏差,与 M 方法偏差的比较。
因为前面说过,转染的质粒中,在 3』UTR 的上游是一个 GFP 蛋白的基因。因此,可以在这里测发光,可以简接地判断上游的蛋白含量的偏差,与 3』UTR 偏差的关联性。
从图中,我们可以看到,大体上,这些点都是顺着斜向的这条线排列的,皮尔森相关系数是 0.81,而且,P 值是 0.00046
把上述的证据整合起来,可以提示,M 方法提示的 RNA 丰度,对于说明表型的水平,是有意义的。
实验内容的第二部分,M 方法灵敏地检测 3』UTR 调节器和功能序列变体

作者分析了所有的寡核苷酸的序列,发现 GC 含量与表达减少效应正相关。
我们看右边的图,图中横坐标是 GC 的百分含量,纵轴是表达倍数变化,图中,我们可以看到这条斜向右下方的线,是这许多个点的趋势线,我们可以从这个线的趋势看出来,GC 含量越高,表达的量越少。
再看左边的这张图,这张图,是最小自由能和表达变化关系的图,我们可以看到,横轴是最小自由能,纵轴是表达变化的倍数。
斜线方向是斜向右上方,这说明最小自由能越大,表达变化的倍数也越大。
而自由能,也就是体现了核酸链的二级结构,也就是说核酸链的二级结构越紧,则表达变化的倍数越小

在更加精细化的分析中,可以看到一些在经验中已知的蛋白结合的基序,和一些被预测过的 miRNA 的基序的作用。
在这里,可以看到 AU 富集的因素,会让表达有减少效应。
经典的 Pumilio 基序,会让表达有减少效应;
miRNA 基序,会让表达有减少效应;
相反,CU 富集的因素,会让表达有增加效应;
而扰乱这些预测元素的变异消除了功能效应。
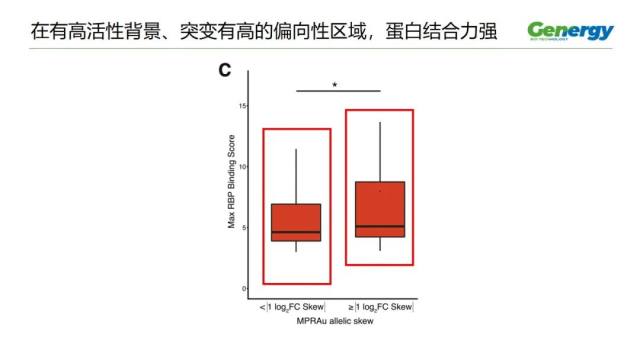
作者还发现,在有高的活性背景、且突变有高的偏向性的区域,蛋白结合力强。
这张图,是一张四分位图,横轴,是突变引起的倍数变化的大小;
纵轴,是最大的红细胞蛋白结合力得分。
我们可以看到,左边引起倍数变化小的,它的 RNA 结合蛋白的结合力也越弱;
右边引起倍数变化大的,它的 RNA 结合蛋白的结合力也更强。
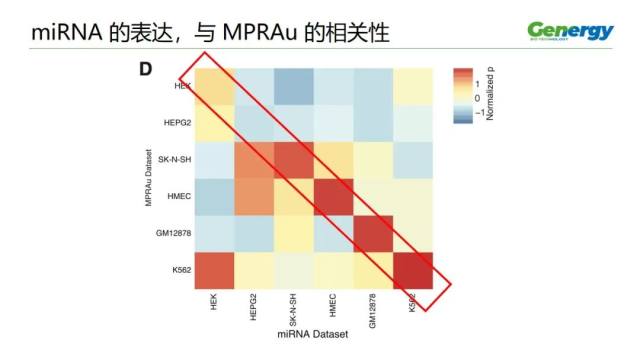
这是每一种细胞中,表达最高的 10 种 miRNA 与这个细胞中的 MPRAu 的相关性,做的交叉比对。
可以明显地看到,大体上,从左上到右下的对角线上的格子的颜色偏红,也就是说,一个细胞中的 M 值会与这个细胞中的 miRNA 有对应关系。
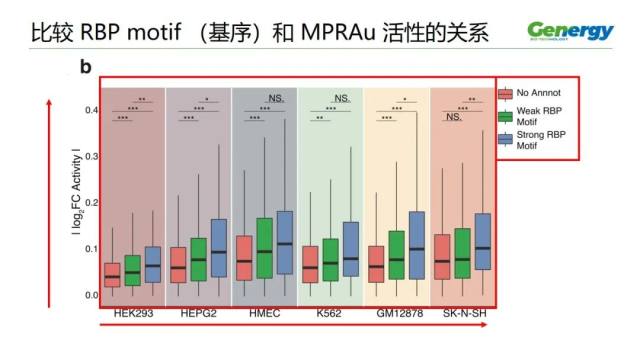
图中,四分位图的三位种颜色,分别对应于 3』UTR 中是否有强的 RNA 结合蛋白的结合位点的基序。红色是没有被注释的基序的基因,绿色是有弱的被注释的基序的基因,蓝色是有强的被注释的基序的基因。
图中,横轴上从左到右排列着 6 种细胞,
纵轴上排列的是 M 值的改变倍数的 log 值。
大家可以明显地看到,6 种细胞中,都是红的四分位图最矮,蓝色的最高,绿色的居中,也就是说,与 RNA 结合蛋白的目标基序有更多重合的基因,它所产生的 mRNA 数量就更大程度受到扰动实验的影响。改动目标基序,会让 mRNA 数量增加
实验内容的第 3 部分,计算建模揭示了 3』UTR 调控的特征

在确定了 M 方法转录水平背后的关键 3『UTR 特征后,作者训练了作者的测试序列的预测模型,并比较了几种分类模型的灵敏度和特异性,以预测具有减弱活性的 3』UTR 元件。
作者最.好的模型在所有细胞类型中都表现良好,
左图,召回率的平均精度为 0.23-0.48
右图,受试者工作特征曲线下面积为 0.67-0.79
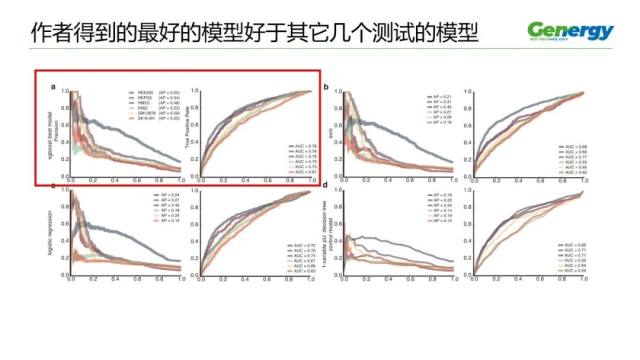
作者得到的最.好的模型,也就是左上角这个用 xgboost 方法得到的模型,好于其它几个测试的模型,也就是好于 b\c\d 这几个图中的模型
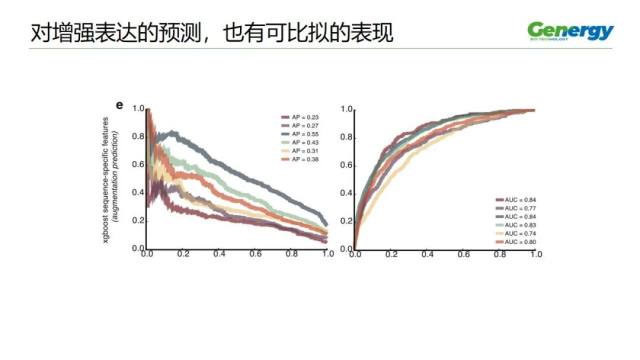
作者还把相同的特征用于预测增强表达,结果发现可以比拟的表现。

接着,作者进一步做了加入 miRNA 和 RBP 的因素进行分析,结果发现加入这两个因素,并不能让预测结果变得更准。
这提示,前面的简单的特征对预测已经足够好。

有几个特征对于预测很重要。
包括 homopolyer,也就是同聚物的长度,,序列的多样性和 U 碱基相关的序列,例如:U/UC,UA/UU 的双碱基的数量

作者发现,最低自由能,也就是 mfe,和减少的预期单调地负相关。
令人惊讶的是,作者 发现尿嘧啶含量的比例对衰减具有非线性影响,低尿嘧啶含量和高尿嘧啶含量均显示衰减效应。具体而言,较长的尿嘧啶均聚物表现出最大的衰减活性
实验内容的第四部分,MPRAu 等位基因效应反映在基因表达和人类表型变化中

在证明了 M 方法检测 3『UTR 元件活性的能力后,作者调查了自己的 tamVar 等位基因效应是否受到改变英国生物银行捕获的转录输出和/或表型特征的因果等位基因的支持。
作者将 GM12878 中的 tamVars 与 Geuvadis RNA 测序 (RNA-seq) 数据集中杂合个体的细胞类型匹配等位基因特异性表达 (ASE) 数据进行了比较,并使用这种比较来估计阳性预测值( PPV) 用于测定中的 tamVar。
作者观察到 tamVars 和内源性观察到的 ASE 之间有中等强的一致性,66.1% 方向一致性,也就是一致的有 42 个,不一致的有 21 个,而二项式 p = 0.011),对应于 32% 的 PPV。

随着用更严格的 ASE 调用(两侧 t 检验 p < 0.001)(STAR 方法),方向性的一致性增加到 77.5%(二项式 p = 6.8 * 10^-4;PPV 为 55%)。
当与 Geuvadis 表达数量性状基因座 (eQTL) 数据重叠时,作者获得了较弱的一致性(方向性一致性为 60.5%,二项式 p = 0.22,PPV 为 20.9%)(图 S5A),这可能是由于不同的调控因素(即 RBP/ miRNA 浓度)在个体间聚集时会减弱真正的等位基因效应

接下来,作者扩展了他的分析,将 tamVars 与来自 GTEx Consortium,2020 年的组织 eQTL 进行比较,作者从遗传精细定位中获得了推定的因果等位基因。
聚合跨细胞类型和组织的等位基因效应,作者观察到具有高推断因果关系概率的变体在聚合 MPRAu 和 GTEx 中值效应大小之间的方向性上显示出显着一致

接下来,作者在英国生物银行中寻找与 94 个性状相关的遗传精细定位的因果变异中 M 方法 tamVar 的富集。作者观察到随着因果关系 (PIP) 阈值的增加,M 值功能更加丰富。这表明除了引起体内基因表达变化外,我们研究中鉴定的 tamVars 还具有表型后果,并且 M 方法是剖析关联研究的有力方法。
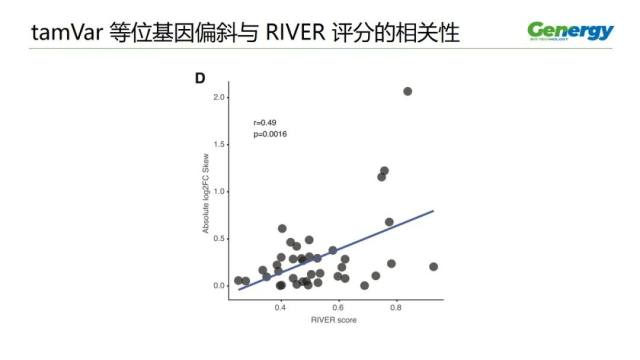
作为确认由 MPRAu 鉴定的 tamVar 与体内表达变化相关的正交方法,作者还分析了一组与大转录效应相关的罕见变异。
当作者将 MPRAu 等位基因偏斜与罕见的变异功能指标 (RIVER 评分)进行比较时,作者观察到显着的正相关。这一发现表明 MPRAu 可以识别共同的功能以及罕见的 3』UTR 变体,现代关联研究对这些变体的检测能力较低
实验内容的第五部分,MPRAu SNV 和删除平铺剖析功能序列基序


在 rs16975240 这个位点,
如果是参考基因序列,那么平均的减少程度很强,它的减少倍数的 log2 的值是-2.29
而如果是变异基因序列,那么减少程度就几乎很少了,它的减少倍数的 log2 的值是-0.24.
在参考序列的这个 SNP 位置的缺失 5 个碱基,或者在它的上游的 10 个 bp 范围内缺失 5 个碱基,减少程度会缓解 3.55 倍到 4.46 倍。
而在变异序列的这个 SNP 位置的相同位置的缺失,缓解程度很小,只有 1.13 倍到 1.16 倍。

这是对这个位点进一步做的逐个碱基的分析。
左边是对参考序列做分析,右边是对变异的序列做的分析。
左边的图中,显示出了每个碱基位置,哪一种碱基是最高富集得分。
可以清楚地看到,从-8 到+1,都出现了高度富集的碱基。
再看右边的对变异的序列做分析的图,
右边的图中,只在-7 的位置,A 碱基是有富集的;
在 0 的位置,U 碱基是有富集的。
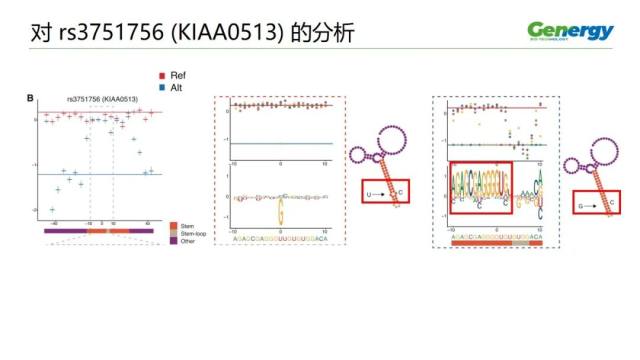
这是对 rs3751756 这个位点的分析。
这个位点有很大的等位基因偏向。
从两个茎环的结构图中,我们可以看到,在这个位置,如果是 U 碱基,就会形成一个突起;
如果是 G 碱基,就会形成稳定的双链,而双链结构能促进与 RNA 结合蛋白的结合。
而且,在茎环结构上每一个碱基的突变,都会有很大的扰动作用。
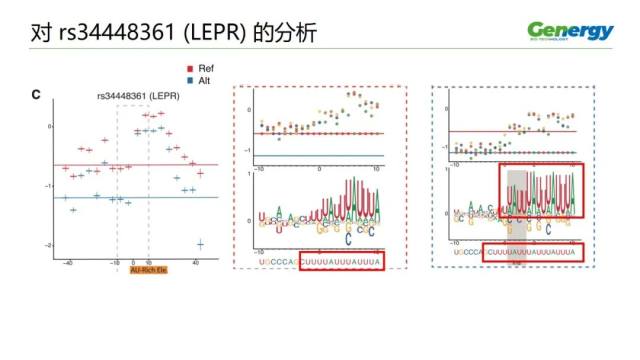
第三个例子是 rs34448361,这个位点,我们可以看它的序列,
它的参考序列是一连串的 AUUUA,AUUUA 的序列重复了 4 次的。
而变异的序列是加入了第 5 个 AUUUA 序列。而且加入的这第 5 个 AUUUA 序列会在很大程度上强化缩减效应。
SNV tiling 再现了这个发现,凡打破 AUUUA 序列的变异,都会破坏缩减效应。
这个 SNP 位点,在 LEPR 基因上,而 LEPR 这个基因与控制体重、吃饭的饱饿感有关,还与现代人、古代人适应冷的环境有关。
实验内容的第六部分,MPRAu 识别与人类进化和疾病相关的因果 3』UTR 变异

在已经用 M 方法确认了前面所说的这些 SNP 的确会影响到 mRNA 的含量,那么接下来就是要把这个方法应用到疾病中去,看如何来确认 3』UTR 的 SNP 如何影响发病。
在 2153 个 GWAS 相关的 3』UTR 中,作者发现了 677 个位点是在至少一种细胞中有显著性差异的。
作者在文章中举了一些例子。这个例子是 rs705866 这个 SNP 位点。
Rs705866 这个位点,落在 PILRB 这个基因上,这个基因与老年黄斑退化相关。
这个 SNP 位点离 rs7803454 很近。而 Rs7803454 在 GWAS 中,是一个标签 SNP 位点。
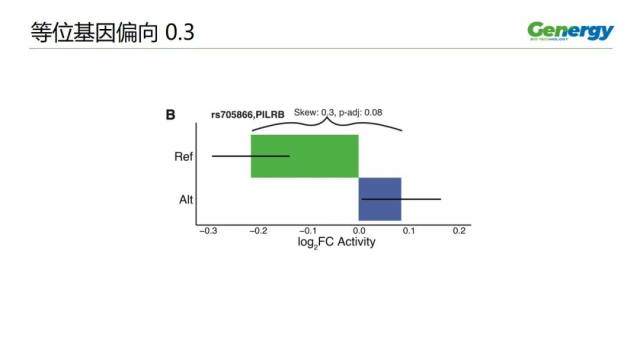
用 M 方法,测到这个 SNP 位点造成的等位基因偏向达到 0.3

为了证实这个 SNP 位点的作用,作者用 CRISPR 方法在神经细胞 SK-N-SH 细胞中做基因改变,
把原来的细胞中的参考序列的 T 碱基,改到变异后的 C 碱基。
基因改造之后,得到三种细胞,
下面蓝色的,是被按照目标改造的细胞。
中间绿色的,是没有被基因改造拗胞。
上面橙色的,是没有被精确地改造,而变成了有更大的变化细胞。
这三组细胞都被分析,再做比较
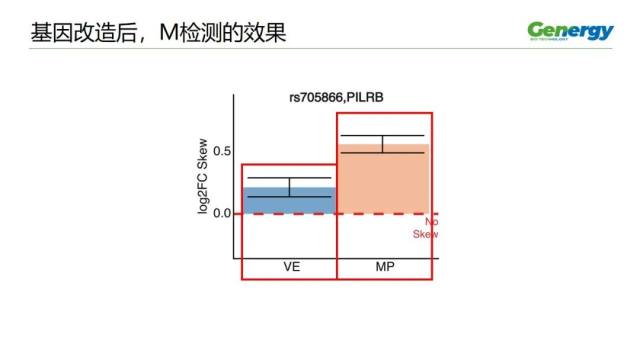
这是基因改造后的 M 检测效果,
可以看到,蓝色的,按照目的改造的细胞,它的变化是 0.21
而被非精确地、更大改造的细胞,它的变化达到了 0.56.
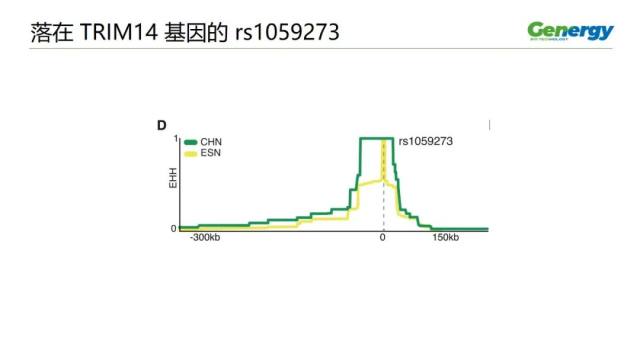
这是落在 TRIM14 基因的编号为 rs1059273 的 SNP 位点。
这个 SNP 在中国的汉人中有富集。而这个 TRIM14 基因与免疫、抗微生物感染有很大的关系。
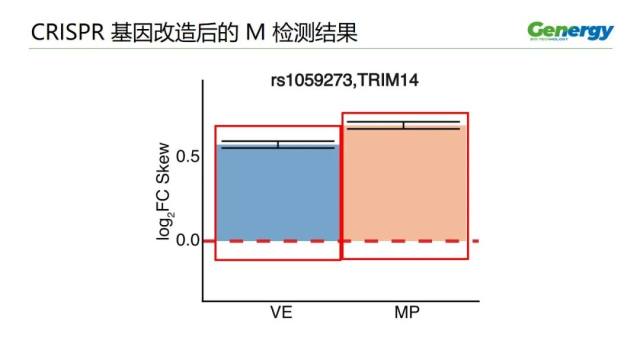
这是对这个位点做 CRISPR 改造后,做 M 检测的结果,
可以看到,按目标进行了基因改造的细胞,偏差值是 0.58;有基因改造,
但不是精确地按目标改造的,偏差值是 0.69
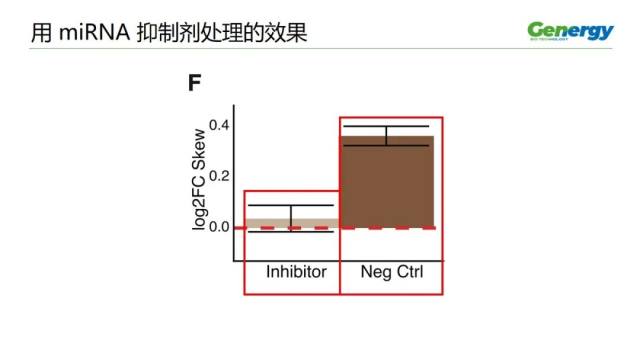
接着,作者用 miRNA 抑制剂进行处理,发现按目标改造的细胞,消除了等位基因偏差;而阴性对照,没有观察到这种情况。
这对于证明 hsa-miR-142-3p 的作用机制,提供了额外的证据。
总结





评论