人类基因组计划完成后,随着测序技术尤其是高通量测序技术的迅速发展,人们对基因组变异与表型之间关系的认识越来越深刻。然而,迄今为止几乎所有测序项目所使用的材料都是组织或一大群细胞的混合物,因而对其研究得到的结果只是大量细胞的平均数据,或者只是这群细胞的代表性信息,单个细胞特异性的信息往往被掩盖了。在基因表达层面上,不同的细胞具有独特的转录组,即便是那些看似相同的细胞群,细胞之间 的 RNA 水平上却差异巨大 [1]。从理论上讲转录组分析应该在单细胞水平上开展,然而由于受到检测的敏感度等技术限制,大部分转录组的研究还只能在几十万或上百万个细胞水平上才能开展。计算出的基因表达水平只是一群细胞的平均值,细胞特异性表达的基因或是某些转录本的剪接体无法被发现。单细胞转录组分析还可用于鉴别两等位基因间的单核苷酸多态性(SNP),用以分析单个细胞中等位基因特异性的基因表达 [2]。
2011 年,《Nature Methods》将单细胞测序列为当年度值得期待的技术之一 [3];2013 年 1 月,《Science》杂志将单细胞测序列为年度最值得关注的六大领域榜首 [4];2014 年 1 月,《Nature Methods》将单细胞测序列为 2013 年度最重要的方法学进展,并且指出刊登在《Nature》系列杂志上的单细胞测序文章在 2013 年出现了大爆发 [5]。目前,单细胞测序已广泛应用于肿瘤细胞异质性研究、新突变位点的发现、肿瘤细胞克隆进化机制及相关新生物标记的鉴定。其中,应用于稀少肿瘤细胞(CTC 细胞和扩散性肿瘤细胞)的早期发现和监控,有助于肿瘤的个性化和精准治疗。

单细胞分离技术
单细胞测序面临的首要问题是如何分离单个目的细胞,目前,主要的单细胞分离方法有有限稀释法、显微操作法、流式细胞法。有限稀释法是一种传统的手段,主要是通过稀释,使某一体积的悬液中理论上只含有一个细胞,然后用移液器吸取相应体积的细胞悬液,便获得了单细胞。这一方法无需特殊设备,目前仍被广泛应用,但该方法依赖于梯度稀释计算,并不是直观下的单细胞分离,因此实际操作中的效率只有 20% 左右,存在大量的空白或多细胞。相比而言,显微操作法是一种能够直接观察到单细胞的方法。这一方法能够准确控制单个细胞的吸取与释放,但是对实验操作的要求较高,不利于规模化操作。而荧光激活细胞流式分选(Fluorescence activated cell sorting,FACS)则能够大规模获得单细胞样本,该方法是利用流式细胞仪,借助于细胞表面标记分选出特定群体的细胞或单个细胞。虽然这一方法需要专门的设备,且细胞的消化和分选过程会对细胞状态产生一定的影响,但是该方法的效率和准确率较高,标准统一,有利于不同实验室实验研究的比较 [6]。

有时候我们可能需要根据样本的具体情况,结合以上多种方法来获取单个细胞。另外,一些新方法的出现也为单细胞基因组学研究提供了便利。意大利 Silicon Biosystems 公司的 DEPArray 系统,利用非均匀电场对细胞施力,并结合形态学的信息,能够从复杂的异质样品中鉴定和捕获单个细胞,整个过程无物理接触或摩擦,保持了细胞活性,并且与各种细胞悬液兼容,也能处理穿刺和组织活检样本 [7]。美国的 Fluidigm 公司的 C1 单细胞全自动制备系统虽然不能对单细胞成像,但大幅提高了自动化水平,该系统基于创新的微流体技术,首先自动分离出 96 个单细胞,随后,在芯片上进行细胞裂解、RNA 反转录及预扩增,整个过程的实现了从单细胞制备到 cDNA 合成放大的全面自动化和规模化 [7]。但分离在细胞群体中罕见(<1%)的单细胞比如 CTC,仍是一项艰巨的挑战 [8]。
对于转录水平的分析,不仅要获得单细胞,同时需尽可能减小对细胞状态的影响。最近,更有学者怀疑非自然的条件下 “推断出的生物学”,因为大多数有关基因表达变化的知识都从培养的细胞所开展研究中获得的。因此,他们开发出了一种史无前例的方法——体内转录组分析(Transcriptome in vivo analysis,TIVA)[9],可以捕获来自活体组织单个细胞中的 mRNA,并且不对周围的组织造成损伤。

单细胞扩增技术
最早的方法出现在 2009 年,即 Tang’s method [1]。Quartz-Seq 的方法,实际上是对 Tang 的方法进行了优化,进一步减少了扩增副产物的产生,同时也简化了实验流程 [10]。CEL-seq 是用 IVT 代替 PCR 达到扩增的目的。基于 SMART 的第一种方法叫做(Single-cell tagged reverse transcription,STRT),但只有转录本的 5’ 端才能被测序 [11]。后来出现的 Smart-Seq [12] 和 Smart-Seq2 [13] 能够使转录本的全长都被测序,而且已经有了较为成熟的商品化试剂盒,是目前应用较多的手段。

单细胞 RNA 测序的应用方向
(1)肿瘤异质性
在基因表达层面上,不同的肿瘤细胞具有独特的转录组,即便是那些看似相同的细胞群,RNA 水平上也可能差异巨大。有研究者利用流式细胞法,对来自 5 位患者的脑胶质瘤细胞进行分选,对获得的单细胞进行 RNA-seq 检测,并深入分析了共 430 例细胞的数据,精细绘制了肿瘤细胞的组成图像。揭示每个胶质母细胞瘤都包含来自多种癌症亚型的细胞,肿瘤之间这些细胞的分布各不相同 [14]。一项黑色素瘤的研究对 19 名病人的单细胞样本进行测序,共计 4645 个不同的肿瘤细胞;类型包括了恶性肿瘤细胞,免疫细胞,间质细胞和内皮细胞。发现在同种肿瘤中的恶性细胞,其转录的异质性与细胞周期,空间分布和抗药性相关 [15]。
(2)发现罕见的细胞类别
传统发现和分离细胞亚群的方法是基于几个已知的蛋白或核酸标志物实现的。但是对于一些罕见的细胞类型来说,鉴定出它们特定的标记目前仍存在很大挑战。利用单细胞 RNA 测序结合一种新的计算方法,研究人员发现了小肠中一些罕见的细胞类型,这对于深入了解器官的细胞组成,探究健康和疾病状态下的组织生物学具有重要意义 [16]。这项成果对于肿瘤研究来说,也许能够用于发现一些罕见的肿瘤细胞类型,如肿瘤干细胞或循环肿瘤细胞。
(3)肿瘤细胞耐药性机制研究及治疗
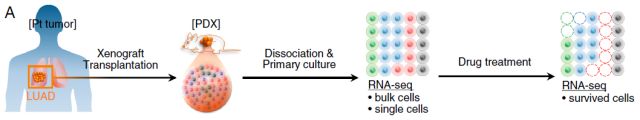
单细胞 RNA 测序在获得表达谱的同时,也能够对转录本上所携带的 SNV 进行分析。例如一篇抗乳腺研究的报道,Paclitaxel(Taxol)是一种常用的癌药物,阻断微管形成,遏制有丝分裂。研究通过单细胞 RNA 测序比较了给药前后的单细胞所携带的 SNV,在单个细胞中检测到了许多细胞群体测序所无法检出的 SNV,并且鉴定出一些耐药细胞特异性的 SNV,与微管形成和稳定,以及细胞粘附和细胞表面信号转导有关 [17]。在肺癌中,研究者利用 PDX 模型,通过单细胞 RNA 测序比较了给药前后的表达谱,聚类分析将细胞分成 4 个分组,并最终找到与耐药相关的细胞 [18]。
另外,对单个 CTC 进行分析也是揭示肿瘤耐药、复发机制的重要手段,目前已开展了很多研究。例如,利用 Smart-Seq,科学家首次对复发黑色素瘤患者血液中单个 CTC 的基因表达进行了研究 [12]。与原发黑色素瘤相比,一些膜蛋白的表达模式显著改变,其中一些基因的低表达跟肿瘤增殖、扩散相关,还有一些则帮助肿瘤循环细胞避开了机体免疫系统的监控。去势疗法(ADT)是前列腺癌的常见治疗手段,但是癌症复发后 AR(androgen receptor,雄激素受体)抑制剂的疗效因人而异。来自哈佛医学院的研究者通过简单的液体活检,获取 CTC 细胞进行单细胞 RNA 测序分析,揭示了前列腺癌的耐药机制 [19]。研究人员从 13 位出现雄激素抵抗(AR)的患者体内收集了 77 份循环肿瘤细胞,并完成了总 RNA 序列分析。通过比对出现 AR 的患者血样循环肿瘤细胞,与未进行治疗患者样品,发现 AR 抗性组出现了 nc-Wnt 信号通路被激活的现象;而 Wnt 信号通路是众所周知的调控细胞生存,增殖与运动功能的信号途径,从而调控癌细胞耐药机制。

(4)在肿瘤免疫微环境中的研究
肿瘤细胞能够采用不同策略,使人体的免疫系统受到抑制,不能正常杀伤肿瘤细胞,上述过程被称为免疫逃逸。免疫治疗通过激活人体自身的免疫系统,恢复机体正常的抗肿瘤免疫反应,从而控制与清除肿瘤。其中抗程序性死亡蛋白 1(programmed death 1, PD-1)及其配体(PD-L1)抑制剂是目前研究最多,发展最快的一种免疫疗法。在各种肿瘤中,接受 PD-1 /PD-L1 抑制剂单药治疗患者的客观有效率也仅为 10-30%,大部分患者对免疫检查抑制剂并不敏感。随着研究的深入,人们逐渐了解到肿瘤微环境的复杂性和多样性,以及它对免疫治疗的重要影响。肿瘤微环境与肿瘤细胞相互作用,共同介导了肿瘤的免疫耐受,从而影响了免疫治疗的效果。抗肿瘤免疫应答是由众多免疫细胞和分子参与的复杂过程,受到机体复杂而精细的调控,这其中的机制仍有待进一步研究。2017 年 6 月,北京大学张泽民课题组及其合作在 Cell 杂志发表了肝癌 T 细胞图谱研究 [20],为肿瘤免疫的图谱勾画做出了范式,也为今后其他肿瘤开展类似的研究及各类肿瘤免疫的发展提供基础。时隔一年,张泽民课题组在此前研究的基础上,进一步在单细胞水平绘制肺癌 T 细胞免疫图谱 [21]。该项工作是国际上迄今为止规模最大的癌症 T 细胞研究,揭示了肺癌 T 细胞的亚群分类、组织分布特征、肿瘤内群体异质性及药物靶基因表达情况,鉴定了跨组织分布的 T 细胞类群及亚群间潜在的状态转换关系,为肺腺癌提供了新的临床标志物。
(5)在早期胚胎发育和干细胞研究中的应用
人类胚胎发育早期仅能收集微量的胚胎细胞和干细胞,这对于了解控制胚胎发育的基因调控网络是一个难题,利用单细胞 RNA-Seq 技术对转录组进行分析则克服了转录组分析对细胞数量要求的限制,近年来已有多篇相关报道。北京大学汤富酬教授课题组早在 2009 年就对单个小鼠卵裂球的进行 RNA-Seq[1],检测到了比微阵列技术多 75%(5270)的表达基因,并且确定了 1753 个新的可变剪接位点。这种单细胞 mRNA- Seq 检测将大大提高我们对单个细胞在哺乳动物发展中转录复杂性的分析能力, 尤其是胚胎发育早期和干细胞这类在体内罕见的细胞群。该课题组 2010 年的另一文章用单细胞测序更加深入地研究了体外条件下内细胞团(Inner cell mass,ICM)转化为胚胎干细胞(Embryonic stem cells,ESCs)的机制 [22]。2013 年,他们又对 90 个处于不同发育阶段的人类早期胚胎细胞以及 34 个胚胎干细胞进行了详尽的分析 [6],检测出了 22,687 个母源表达基因,其中包括 8,701 条长链非编码 RNAs,相比于以往通过 microarray 检测出的 9,735 个母源基因数量大大增加。研究还发现了 2,733 条新的 lncRNAs,其中许多只在特定的发育阶段表达。此外,实验还检测到 1498 个基因在上胚层(Epiblast,EPI)和体外人类胚胎干细胞间存在差异表达,证实了 EPI 和 ESCs 具有显著不同的转录组,有显示差异性表达,从而解答了人类上胚层和体外干细胞之间的基因表达是否相同这一由来已久的问题。
(6)在神经科学中的应用
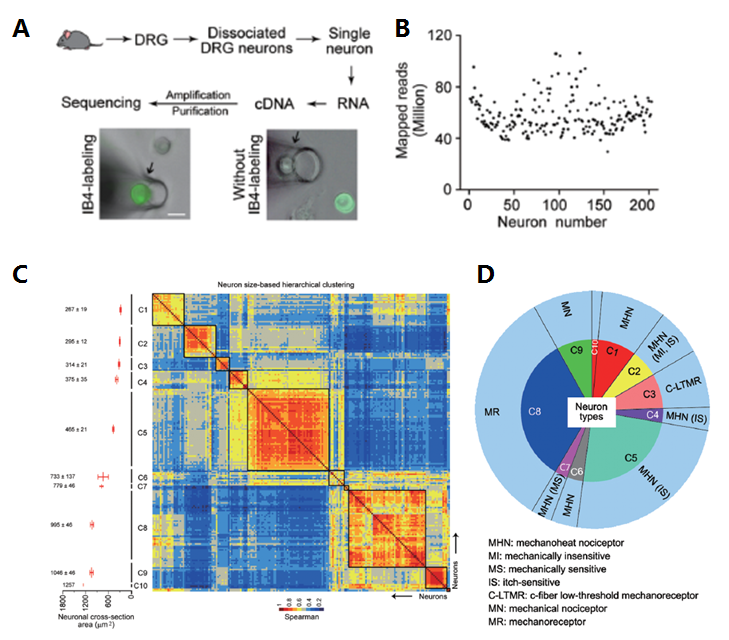
神经科学是单细胞测序的另一个应用领域,哺乳动物的大脑中有数十亿个神经元,每个神经元可根据形态特征,电生理特性,分子标记进行归类。在一群细胞中,单个神经元特异性的信息就被稀释了,少数细胞特异的基因表达则有可能无法检测到。研究不同神经元的表达模式能够为我们提供更全面的基因表达图谱、甚至表达调控网络。而且,将单个神经元的基因表达于神经元的表型信息结合起来,还能帮助我们对神经元进行更加准确和细致的分类。上海伯豪助力中国科学院上海生命科学研究院神经科学研究所张旭院士研究组,通过高覆盖度的单细胞测序和以神经元大小为参考的层次聚类,对小鼠背根神经节初级感觉神经元进行了分类,又通过全细胞膜片钳在体记录结合单细胞 PCR 方法可检测各类初级感觉神经元对外周皮肤刺激的反应。该工作首次通过高覆盖的单细胞测序对初级感觉神经元进行了重新分类,并且建立了基因表达与在体功能的相互关系 [23]。
单细胞多组学测序
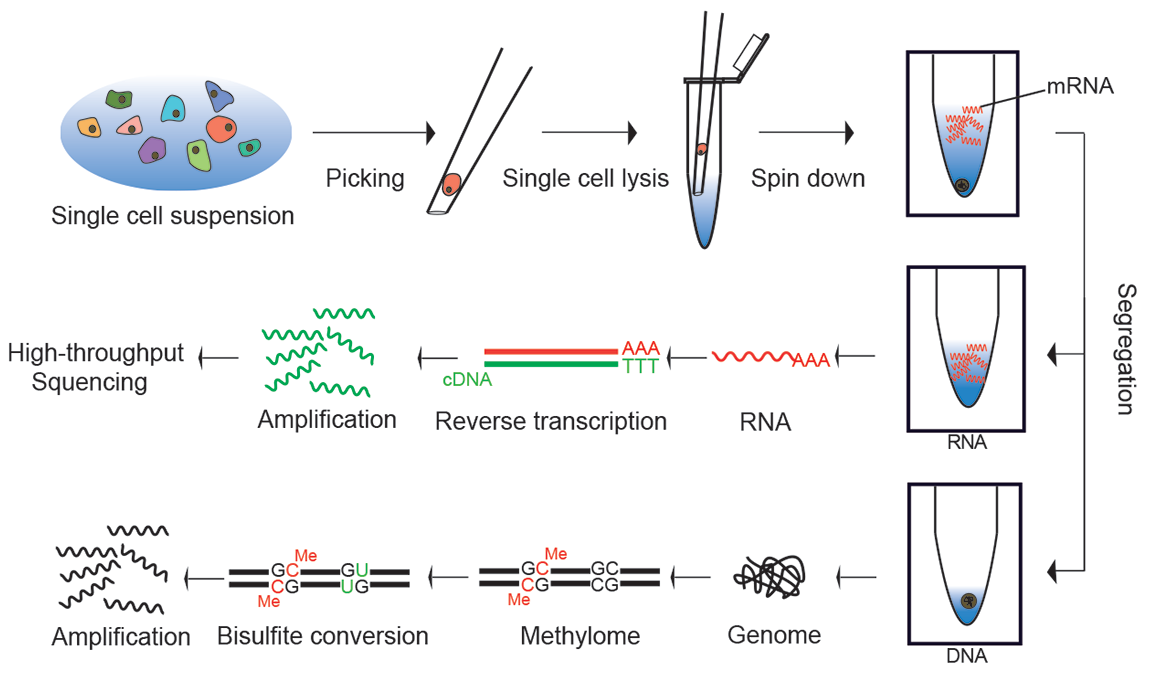
生命现象的发生和调控过程是极其复杂的,在肿瘤等复杂疾病的发生发展过程中,会涉及到基因组、转录组、及表观遗传等多层面的变化及调控。在大数据时代,将多个组学数据结合起来的整合研究——多组学(Multi-omics)研究,是一大趋势。多组学研究可以掌握疾病全局的变化过程,为研究肿瘤调控机制和精准医疗提供综合解决方案。虽然在同一个细胞内对基因组,转录组和甲基化进行测序非常困难,还是有一些方法被开发出来。
(1)单细胞 DNA 和 RNA 平行测序
单细胞 G&T-seq 技术实现了对单个细胞内的 DNA 和 RNA 平行测序 [24],能够展现单个细胞的基因变异与基因功能之间的关系,这个技术可以分析单细胞基因型和表现型之间的关系,深刻揭示一个细胞内的 DNA 信息指导细胞状态的调控机制,真正实现了对同一个细胞在一个时空内遗传物质的综合研究。
(2)单细胞 RNA 和甲基化平行测序
DNA 甲基化对基因的转录水平即 RNA 的表达具有调控作用,scM&T-seq 技术实现了对单个内的 RNA 和 DNA 甲基化进行测序 [25]。同济大学的研究小组通过对同一个细胞的细胞质细胞核分别进行 RNA 测序和 DNA 甲基化测序,也实现了单个细胞的 RNA 和甲基化平行测序 [26]。
(3)单细胞基因组,转录组,甲基化平行测序
国内几个课题组建立了一种全新的单细胞三重组学测序方法(scTrio-seq)[27],在国际上首次从同一个单细胞中实现对三种组学高通量测序信息的同时获取,并从单细胞水平发现肝癌细胞在三种组学上存在密切相互关联的高度异质性。利用这项技术,对一名肝细胞肝癌病人癌组织中的 25 个癌细胞进行了三种组学的同时分析。三种组学的数据都表明这 25 个肝癌细胞来自两个不同的细胞亚群。进一步分析两个亚群在三种组学上的差异发现,在被检测肝癌细胞中,数量上占比较小的亚群 I 细胞拥有更多的 DNA 拷贝数变异以及更高的 DNA 甲基化水平,可能更易逃脱患者免疫应答系统的识别,因而可能更加容易发生转移。
大规模低成本的单细胞测序方法
单细胞测序目前存在的一个很大问题的成本较高,尤其在需要最大规模的细胞进行测序时。虽然测序本身的成本已经很低,但因为每个细胞的扩增、文库构建都需要单独进行,这部分的试剂成本非常高昂。
哈佛大学医学院的研究人员开发了以微滴为基础的两种独立技术 Drop-seq 和 inDrop。他们利用微流体装置将微珠(扩增引物固定在微珠上,并且引物带有用于识别细胞来源的条码)和细胞一起装入微小的液滴,建立了快速、廉价、高通量的单细胞 RNA-seq 方法。由此并行检测数以千计的细胞。研究者们认为,Drop-seq [28] 和 inDrop [29] 能够帮助科学家进一步对发人体细胞进行分类,绘制复杂组织的细胞多样性图谱。类似的技术还有 SCRB-Seq [30],CytoSeq [31] 等。

最近,一项最新研究成果为单细胞测序技术带来了突破,有望将单个细胞的测序成本降至 1 美分。SPLiT-seq 方法能兼容与甲醛固定的细胞或细胞核,且无需昂贵的专用设备,省去用定制化微流体或其他微孔分选单细胞的过程,而是用该细胞当作其自身 RNA 的隔离室。将细胞池分成多个组,引入成本低廉的组合条形码。SPLiT-seq 技术最关键的实现过程,其实就类似于一个 “洗牌” 的过程,通过这四次洗牌步骤,理论上可以得到 21,233,664 种条形码组合(在 96 孔板中进行三轮条形码编码,然后进行第四轮 PCR 反应,可以特异性的标记超过 100 万个细胞),最终给每个细胞一个独特的组合条形码 [33]。
技术难点及需要解决的问题
单细胞的获取:到目前为止,绝大多数单细胞转录组研究时还是需要单细胞悬液(比如组织解离液或者细胞培养悬液)做检测样品,细胞在解离、分选过程中,其转录状态是否发生改变,是要特别注意的一个问题。随之而来的一个问题是这种样品不能反映细胞在组织里的空间组织结构信息,除非我们知道这些细胞取自组织的哪一个部位。此外,各种各样的细胞构成了一个复杂的系统,进而形成了组织和器官的整体功能,解离状态的细胞打破这种整体性,因此我们还需要进一步发展微环境系统下单个细胞转录组的研究。
单细胞的 RNA 扩增:由于单个细胞里含有的 RNA 非常少,单细胞转录组研究工作主要依赖逆转录反应和扩增,这些反应非常容易出错或者丢失信息,以致产生了大量的偏差。大部分科研人员都会尽量减少 PCR 的反应循环数,就是为了减少这方面造成的误差,此外线性扩增技术还是能够在一定程度上解决这种因为扩增而带来的误差问题。
各种扩增技术带来的另一个不同是信息的保留,比如可以全长对转录本进行扩增和测序,这样能够整个基因及其不同转录异构体(transcript isoform)的结构信息,而我们也可以采取只对转录本的 5' 或 3' 端进行扩增的策略,只保留基因表达丰度的信息。再比如大部分扩增和建库过程都会丢失转录的方向信息,而这对基因表达定量的准确性,反义转录本以及其他非编码 RNA 的研究来说至关重要。最后,非编码 RNA 信息的保留也可能成为今后扩增方法改进的目标。
展望
近几年利用单细胞测序技术,在肿瘤异质性、免疫微环境、神经科学、胚胎发育、细胞分化等研究得到了优异的成果。但是,单细胞测序相关研究中还处在非常初级的阶段,同时也面临很多挑战。技术的不断改进和突变,将使单细胞测序技术在未来疾病精准研究与治疗具有十分广阔的应用前景。
伯豪生物经过 6 年的积累,建立了包括 SMART-seq,10X Genomics 和 BD Rhapsody 的单细胞 RNA 测序平台,为客户提供从样本保存、运输、单细胞悬液制备,到单细胞分选、建库和数据分析的一站式解决方案。目前已为来源于人、小鼠、大鼠、果蝇、斑马鱼、牛、羊等物种的临床和科研样本提供了单细胞测序和数据分析服务,研究成果发表在 Cell Research、Nature Neuroscience 等杂志上。伯豪生物将不断完善技术方法和服务体系,助力客户发表高水平研究论文。
伯豪单细胞测序数据分析展示







[1] Tang F, Barbacioru C, Wang Y, Nordman E, Lee C, et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat Methods 2009, 6(5):377-382.
[2] Deng Q, Ramskold D, Reinius B, Sandberg R. Single-cell RNA-seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science, 343(6167):193-196.
[3] de Souza N. Single-cell genetics. Nat Methods 2013, 10(9):820.
[4] The biology of genomes. Single-cell sequencing tackles basic and biomedical questions. Science 2012, 336(6084):976-7.
[5] Method of the Year 2013. Nat Methods 2014, 11(1):1.
[6] Tang F, Lao K, Surani MA. Development and applications of single-cell transcriptome analysis. Nat Methods 2011, 8(4 Suppl):S6-11.
[7] Smith C. Cancer shows strength through diversity. Nature 2013, 499(7459):505-8.
[8] Harouaka R, Kang Z, Zheng SY, Cao L. Circulating tumor cells: advances in isolation and analysis, and challenges for clinical applications. Pharmacol Ther 2014, 141(2):209-21.
[9] Lovatt D, Ruble BK, Lee J, Dueck H, Kim TK, et al. Transcriptome in vivo analysis (TIVA) of spatially defined single cells in live tissue. Nat Methods 2014, 11(2):190-196.
[10] Sasagawa Y, Nikaido I, Hayashi T, Danno H, Uno KD, et al. Quartz-Seq: a highly reproducible and sensitive single-cell RNA sequencing method, reveals non-genetic gene-expression heterogeneity. Genome Biol 2013, 14(4):R31.
[11] Islam S1, Kjällquist U, Moliner A, Zajac P, Fan JB, et al. Characterization of the single-cell transcriptional landscape by highly multiplex RNA-seq. Genome Res 2011, 21(7):1160-7.
[12] Ramskold D, Luo S, Wang YC, Li R, Deng Q, et al. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat Biotechnol 2012, 30(8):777-782.
[13] Picelli S, Faridani OR, Björklund AK, Winberg G, Sagasser S, et al. Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc 2014, 9(1):171-81.
[14] Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344(6190):1396-401.
[15] Tirosh I, Izar B, Prakadan SM, Wadsworth MH, Treacy D, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352(6282):189-96.
[16] Grün D, Lyubimova A, Kester L, Wiebrands K, Basak O, et al. Single-cell messenger RNA sequencing reveals rare intestinal cell types. Nature 2015, 525(7568):251-5.
[17] Lee MC, Lopez-Diaz FJ, Khan SY, Tariq MA, Dayn Y, et al. Single-cell analyses of transcriptional heterogeneity during drug tolerance transition in cancer cells by RNA sequencing. Proc Natl Acad Sci U S A 2014, 111(44):E4726-35.
[18 Kim KT, Lee HW, Lee HO, Kim SC, Seo YJ, et al. Single-cell mRNA sequencing identifies subclonal heterogeneity in anti-cancer drug responses of lung adenocarcinoma cells. Genome Biol 2015, 16:127.
[19] Miyamoto DT, Zheng Y, Wittner BS, Lee RJ, Zhu H, et al. RNA-Seq of single prostate CTCs implicates noncanonical Wnt signaling in antiandrogen resistance. Science 2015, 349(6254):1351-6.
[20] Zheng C, Zheng L, Yoo JK, et al. Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell 2017, 169(7):1342-1356.
[21] Guo X, Zhang Y, Zheng L, et al. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat Med 2018, 24(7):978-985.
[22] Li L, Dong J, Yan L, et al. Single-Cell RNA-Seq Analysis Maps Development of Human Germline Cells and Gonadal Niche Interactions. Cell Stem Cell 2017, 20(6):858-873.
[23] Li CL, Li KC, Wu D, et al. Somatosensory neuron types identified by high-coverage single-cell RNA-sequencing and functional heterogeneity. Cell Res 2015, 26(1):83-102.
[24] Macaulay IC, Haerty W, Kumar P, et al. G&T-seq: parallel sequencing of single-cell genomes andtranscriptomes. Nature methods 2015, 12(6): 519-522.
[25] Angermueller C, Clark SJ, Lee HJ, Macaulay IC, Teng MJ, et al. Reik Parallel single-cell sequencing links transcriptional and epigenetic heterogeneity. Nat Methods 2016, 13(3):229-32.
[26] Hu Y, Huang K, An Q, Du G, Hu G, et al. Simultaneous profiling of transcriptome and DNA methylome from a single cell. Genome Biol 2016, 17:88.
[27] Hou Y, Guo H, Cao C, Li X, Hu B, et al. Single-cell triple omics sequencing reveals genetic, epigenetic, and transcriptomic heterogeneity in hepatocellular carcinomas. Cell Res 2016, 26(3):304-19.
[28] Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015, 161(5):1202-14.
[29] Klein AM, Mazutis L, Akartuna I, Tallapragada N, Veres A, et al. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 2015, 161(5):1187-201.
[30] M Soumillon, D Cacchiarelli, S Semrau, et al. Characterization of directed differentiation by high-throughput single-cell RNA-Seq. Anatomical Record 2014 , 217(1):16
[31] Fan HC1, Fu GK1, Fodor SP2. Expression profiling. Combinatorial labeling of single cells for gene expression cytometry. Science 2015, 347(6222):1258367.
[32] Zheng GX, Terry JM, Belgrader P, Ryvkin P, Bent ZW, et al. Montesclaros Massively parallel digital transcriptional profiling of single cells. Nat Commun 2017, 8:14049.
[33] Rosenberg AB, Roco CM, Muscat RA,Kuchina A, Sample P, et al. Single-cell profiling of the developing mouse brainand spinal cord with split-pool barcoding. Science 2018, 360(6385):176-182.

2011 年,《Nature Methods》将单细胞测序列为当年度值得期待的技术之一 [3];2013 年 1 月,《Science》杂志将单细胞测序列为年度最值得关注的六大领域榜首 [4];2014 年 1 月,《Nature Methods》将单细胞测序列为 2013 年度最重要的方法学进展,并且指出刊登在《Nature》系列杂志上的单细胞测序文章在 2013 年出现了大爆发 [5]。目前,单细胞测序已广泛应用于肿瘤细胞异质性研究、新突变位点的发现、肿瘤细胞克隆进化机制及相关新生物标记的鉴定。其中,应用于稀少肿瘤细胞(CTC 细胞和扩散性肿瘤细胞)的早期发现和监控,有助于肿瘤的个性化和精准治疗。
单细胞分离技术
单细胞测序面临的首要问题是如何分离单个目的细胞,目前,主要的单细胞分离方法有有限稀释法、显微操作法、流式细胞法。有限稀释法是一种传统的手段,主要是通过稀释,使某一体积的悬液中理论上只含有一个细胞,然后用移液器吸取相应体积的细胞悬液,便获得了单细胞。这一方法无需特殊设备,目前仍被广泛应用,但该方法依赖于梯度稀释计算,并不是直观下的单细胞分离,因此实际操作中的效率只有 20% 左右,存在大量的空白或多细胞。相比而言,显微操作法是一种能够直接观察到单细胞的方法。这一方法能够准确控制单个细胞的吸取与释放,但是对实验操作的要求较高,不利于规模化操作。而荧光激活细胞流式分选(Fluorescence activated cell sorting,FACS)则能够大规模获得单细胞样本,该方法是利用流式细胞仪,借助于细胞表面标记分选出特定群体的细胞或单个细胞。虽然这一方法需要专门的设备,且细胞的消化和分选过程会对细胞状态产生一定的影响,但是该方法的效率和准确率较高,标准统一,有利于不同实验室实验研究的比较 [6]。
对于转录水平的分析,不仅要获得单细胞,同时需尽可能减小对细胞状态的影响。最近,更有学者怀疑非自然的条件下 “推断出的生物学”,因为大多数有关基因表达变化的知识都从培养的细胞所开展研究中获得的。因此,他们开发出了一种史无前例的方法——体内转录组分析(Transcriptome in vivo analysis,TIVA)[9],可以捕获来自活体组织单个细胞中的 mRNA,并且不对周围的组织造成损伤。
TIVA 技术原理
单细胞扩增技术
最早的方法出现在 2009 年,即 Tang’s method [1]。Quartz-Seq 的方法,实际上是对 Tang 的方法进行了优化,进一步减少了扩增副产物的产生,同时也简化了实验流程 [10]。CEL-seq 是用 IVT 代替 PCR 达到扩增的目的。基于 SMART 的第一种方法叫做(Single-cell tagged reverse transcription,STRT),但只有转录本的 5’ 端才能被测序 [11]。后来出现的 Smart-Seq [12] 和 Smart-Seq2 [13] 能够使转录本的全长都被测序,而且已经有了较为成熟的商品化试剂盒,是目前应用较多的手段。
单细胞 RNA 测序的应用方向
(1)肿瘤异质性
在基因表达层面上,不同的肿瘤细胞具有独特的转录组,即便是那些看似相同的细胞群,RNA 水平上也可能差异巨大。有研究者利用流式细胞法,对来自 5 位患者的脑胶质瘤细胞进行分选,对获得的单细胞进行 RNA-seq 检测,并深入分析了共 430 例细胞的数据,精细绘制了肿瘤细胞的组成图像。揭示每个胶质母细胞瘤都包含来自多种癌症亚型的细胞,肿瘤之间这些细胞的分布各不相同 [14]。一项黑色素瘤的研究对 19 名病人的单细胞样本进行测序,共计 4645 个不同的肿瘤细胞;类型包括了恶性肿瘤细胞,免疫细胞,间质细胞和内皮细胞。发现在同种肿瘤中的恶性细胞,其转录的异质性与细胞周期,空间分布和抗药性相关 [15]。
(2)发现罕见的细胞类别
传统发现和分离细胞亚群的方法是基于几个已知的蛋白或核酸标志物实现的。但是对于一些罕见的细胞类型来说,鉴定出它们特定的标记目前仍存在很大挑战。利用单细胞 RNA 测序结合一种新的计算方法,研究人员发现了小肠中一些罕见的细胞类型,这对于深入了解器官的细胞组成,探究健康和疾病状态下的组织生物学具有重要意义 [16]。这项成果对于肿瘤研究来说,也许能够用于发现一些罕见的肿瘤细胞类型,如肿瘤干细胞或循环肿瘤细胞。
(3)肿瘤细胞耐药性机制研究及治疗
单细胞 RNA 测序在获得表达谱的同时,也能够对转录本上所携带的 SNV 进行分析。例如一篇抗乳腺研究的报道,Paclitaxel(Taxol)是一种常用的癌药物,阻断微管形成,遏制有丝分裂。研究通过单细胞 RNA 测序比较了给药前后的单细胞所携带的 SNV,在单个细胞中检测到了许多细胞群体测序所无法检出的 SNV,并且鉴定出一些耐药细胞特异性的 SNV,与微管形成和稳定,以及细胞粘附和细胞表面信号转导有关 [17]。在肺癌中,研究者利用 PDX 模型,通过单细胞 RNA 测序比较了给药前后的表达谱,聚类分析将细胞分成 4 个分组,并最终找到与耐药相关的细胞 [18]。
肿瘤耐药研究
另外,对单个 CTC 进行分析也是揭示肿瘤耐药、复发机制的重要手段,目前已开展了很多研究。例如,利用 Smart-Seq,科学家首次对复发黑色素瘤患者血液中单个 CTC 的基因表达进行了研究 [12]。与原发黑色素瘤相比,一些膜蛋白的表达模式显著改变,其中一些基因的低表达跟肿瘤增殖、扩散相关,还有一些则帮助肿瘤循环细胞避开了机体免疫系统的监控。去势疗法(ADT)是前列腺癌的常见治疗手段,但是癌症复发后 AR(androgen receptor,雄激素受体)抑制剂的疗效因人而异。来自哈佛医学院的研究者通过简单的液体活检,获取 CTC 细胞进行单细胞 RNA 测序分析,揭示了前列腺癌的耐药机制 [19]。研究人员从 13 位出现雄激素抵抗(AR)的患者体内收集了 77 份循环肿瘤细胞,并完成了总 RNA 序列分析。通过比对出现 AR 的患者血样循环肿瘤细胞,与未进行治疗患者样品,发现 AR 抗性组出现了 nc-Wnt 信号通路被激活的现象;而 Wnt 信号通路是众所周知的调控细胞生存,增殖与运动功能的信号途径,从而调控癌细胞耐药机制。
CTC 单细胞测序
(4)在肿瘤免疫微环境中的研究
肿瘤细胞能够采用不同策略,使人体的免疫系统受到抑制,不能正常杀伤肿瘤细胞,上述过程被称为免疫逃逸。免疫治疗通过激活人体自身的免疫系统,恢复机体正常的抗肿瘤免疫反应,从而控制与清除肿瘤。其中抗程序性死亡蛋白 1(programmed death 1, PD-1)及其配体(PD-L1)抑制剂是目前研究最多,发展最快的一种免疫疗法。在各种肿瘤中,接受 PD-1 /PD-L1 抑制剂单药治疗患者的客观有效率也仅为 10-30%,大部分患者对免疫检查抑制剂并不敏感。随着研究的深入,人们逐渐了解到肿瘤微环境的复杂性和多样性,以及它对免疫治疗的重要影响。肿瘤微环境与肿瘤细胞相互作用,共同介导了肿瘤的免疫耐受,从而影响了免疫治疗的效果。抗肿瘤免疫应答是由众多免疫细胞和分子参与的复杂过程,受到机体复杂而精细的调控,这其中的机制仍有待进一步研究。2017 年 6 月,北京大学张泽民课题组及其合作在 Cell 杂志发表了肝癌 T 细胞图谱研究 [20],为肿瘤免疫的图谱勾画做出了范式,也为今后其他肿瘤开展类似的研究及各类肿瘤免疫的发展提供基础。时隔一年,张泽民课题组在此前研究的基础上,进一步在单细胞水平绘制肺癌 T 细胞免疫图谱 [21]。该项工作是国际上迄今为止规模最大的癌症 T 细胞研究,揭示了肺癌 T 细胞的亚群分类、组织分布特征、肿瘤内群体异质性及药物靶基因表达情况,鉴定了跨组织分布的 T 细胞类群及亚群间潜在的状态转换关系,为肺腺癌提供了新的临床标志物。
(5)在早期胚胎发育和干细胞研究中的应用
人类胚胎发育早期仅能收集微量的胚胎细胞和干细胞,这对于了解控制胚胎发育的基因调控网络是一个难题,利用单细胞 RNA-Seq 技术对转录组进行分析则克服了转录组分析对细胞数量要求的限制,近年来已有多篇相关报道。北京大学汤富酬教授课题组早在 2009 年就对单个小鼠卵裂球的进行 RNA-Seq[1],检测到了比微阵列技术多 75%(5270)的表达基因,并且确定了 1753 个新的可变剪接位点。这种单细胞 mRNA- Seq 检测将大大提高我们对单个细胞在哺乳动物发展中转录复杂性的分析能力, 尤其是胚胎发育早期和干细胞这类在体内罕见的细胞群。该课题组 2010 年的另一文章用单细胞测序更加深入地研究了体外条件下内细胞团(Inner cell mass,ICM)转化为胚胎干细胞(Embryonic stem cells,ESCs)的机制 [22]。2013 年,他们又对 90 个处于不同发育阶段的人类早期胚胎细胞以及 34 个胚胎干细胞进行了详尽的分析 [6],检测出了 22,687 个母源表达基因,其中包括 8,701 条长链非编码 RNAs,相比于以往通过 microarray 检测出的 9,735 个母源基因数量大大增加。研究还发现了 2,733 条新的 lncRNAs,其中许多只在特定的发育阶段表达。此外,实验还检测到 1498 个基因在上胚层(Epiblast,EPI)和体外人类胚胎干细胞间存在差异表达,证实了 EPI 和 ESCs 具有显著不同的转录组,有显示差异性表达,从而解答了人类上胚层和体外干细胞之间的基因表达是否相同这一由来已久的问题。
(6)在神经科学中的应用
神经科学是单细胞测序的另一个应用领域,哺乳动物的大脑中有数十亿个神经元,每个神经元可根据形态特征,电生理特性,分子标记进行归类。在一群细胞中,单个神经元特异性的信息就被稀释了,少数细胞特异的基因表达则有可能无法检测到。研究不同神经元的表达模式能够为我们提供更全面的基因表达图谱、甚至表达调控网络。而且,将单个神经元的基因表达于神经元的表型信息结合起来,还能帮助我们对神经元进行更加准确和细致的分类。上海伯豪助力中国科学院上海生命科学研究院神经科学研究所张旭院士研究组,通过高覆盖度的单细胞测序和以神经元大小为参考的层次聚类,对小鼠背根神经节初级感觉神经元进行了分类,又通过全细胞膜片钳在体记录结合单细胞 PCR 方法可检测各类初级感觉神经元对外周皮肤刺激的反应。该工作首次通过高覆盖的单细胞测序对初级感觉神经元进行了重新分类,并且建立了基因表达与在体功能的相互关系 [23]。
神经元单细胞测序及亚群分类
单细胞多组学测序
生命现象的发生和调控过程是极其复杂的,在肿瘤等复杂疾病的发生发展过程中,会涉及到基因组、转录组、及表观遗传等多层面的变化及调控。在大数据时代,将多个组学数据结合起来的整合研究——多组学(Multi-omics)研究,是一大趋势。多组学研究可以掌握疾病全局的变化过程,为研究肿瘤调控机制和精准医疗提供综合解决方案。虽然在同一个细胞内对基因组,转录组和甲基化进行测序非常困难,还是有一些方法被开发出来。
(1)单细胞 DNA 和 RNA 平行测序
单细胞 G&T-seq 技术实现了对单个细胞内的 DNA 和 RNA 平行测序 [24],能够展现单个细胞的基因变异与基因功能之间的关系,这个技术可以分析单细胞基因型和表现型之间的关系,深刻揭示一个细胞内的 DNA 信息指导细胞状态的调控机制,真正实现了对同一个细胞在一个时空内遗传物质的综合研究。
(2)单细胞 RNA 和甲基化平行测序
DNA 甲基化对基因的转录水平即 RNA 的表达具有调控作用,scM&T-seq 技术实现了对单个内的 RNA 和 DNA 甲基化进行测序 [25]。同济大学的研究小组通过对同一个细胞的细胞质细胞核分别进行 RNA 测序和 DNA 甲基化测序,也实现了单个细胞的 RNA 和甲基化平行测序 [26]。
(3)单细胞基因组,转录组,甲基化平行测序
国内几个课题组建立了一种全新的单细胞三重组学测序方法(scTrio-seq)[27],在国际上首次从同一个单细胞中实现对三种组学高通量测序信息的同时获取,并从单细胞水平发现肝癌细胞在三种组学上存在密切相互关联的高度异质性。利用这项技术,对一名肝细胞肝癌病人癌组织中的 25 个癌细胞进行了三种组学的同时分析。三种组学的数据都表明这 25 个肝癌细胞来自两个不同的细胞亚群。进一步分析两个亚群在三种组学上的差异发现,在被检测肝癌细胞中,数量上占比较小的亚群 I 细胞拥有更多的 DNA 拷贝数变异以及更高的 DNA 甲基化水平,可能更易逃脱患者免疫应答系统的识别,因而可能更加容易发生转移。
大规模低成本的单细胞测序方法
单细胞测序目前存在的一个很大问题的成本较高,尤其在需要最大规模的细胞进行测序时。虽然测序本身的成本已经很低,但因为每个细胞的扩增、文库构建都需要单独进行,这部分的试剂成本非常高昂。
哈佛大学医学院的研究人员开发了以微滴为基础的两种独立技术 Drop-seq 和 inDrop。他们利用微流体装置将微珠(扩增引物固定在微珠上,并且引物带有用于识别细胞来源的条码)和细胞一起装入微小的液滴,建立了快速、廉价、高通量的单细胞 RNA-seq 方法。由此并行检测数以千计的细胞。研究者们认为,Drop-seq [28] 和 inDrop [29] 能够帮助科学家进一步对发人体细胞进行分类,绘制复杂组织的细胞多样性图谱。类似的技术还有 SCRB-Seq [30],CytoSeq [31] 等。
Drop-seq 技术原理
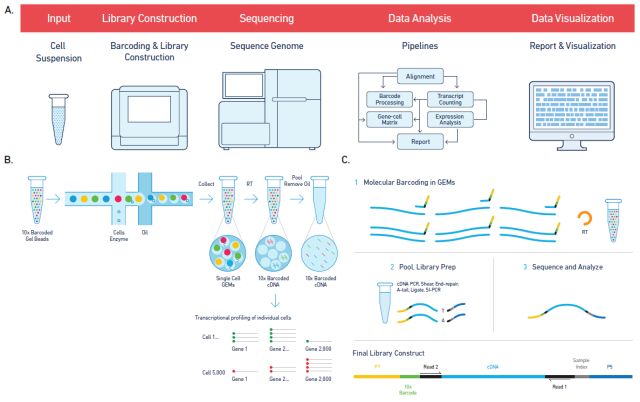
10X genomics 公司的 Chromium 系统,利用微流控技术将细胞自动分配到 100,000s 到 1000,000s 微反应体系,每个微反应体系含有一种特定的 barcode 序列;含有 barcode 信息的凝胶珠子(GEMs)首先与样品和酶的混合物混合,然后与位于微流体 “双十字” 连接中的油表面活性剂溶液结合;GEMs 流到储液器中并进行收集。凝胶珠子溶解释放 barcode 序列,开始对样本进行标记;最后将每个液滴中含有 barcode 信息的产物混合,然后构建标准测序文库。利用这项技术,研究人员从 29 个样本中收集了大约 25 万个单细胞的转录组数据 [32]。结果显示该系统具有相当高的灵敏度,并且有能力鉴别出罕见的细胞亚群。
10X genomics 技术原理
BD Rhapsody 技术利用卡式芯片在磁性的寡核苷酸条形码标记微球上实现单细胞捕获和 mRNA 转录本的分子标签,然后将这些微球合并到单个管中用于 cDNA 扩增和文库构建。可满足 100~10000 个细胞的自动分选、扩增及建库。此外,该技术使用带有寡核苷酸的高质量抗体(Ab-oligos),这条寡核苷酸带有抗体特异的条形码,细胞在经过 Ab-oligos 标记后,可在单细胞水平同时获得转录组和蛋白表达。
BD Rhapsody 实验流程
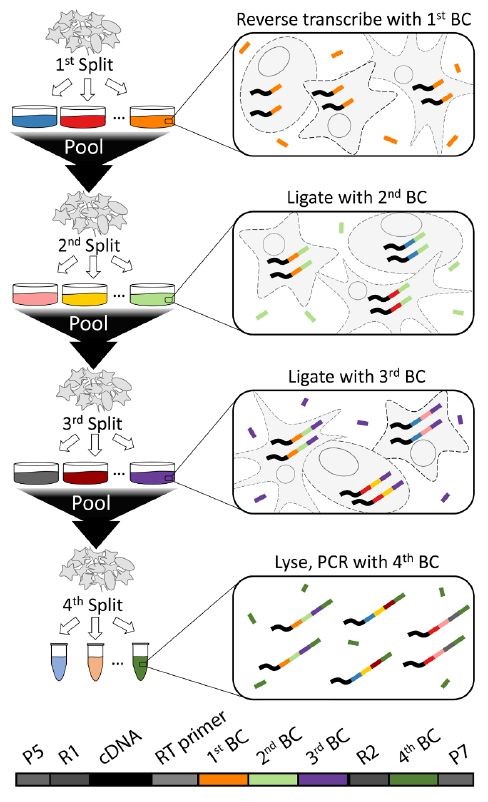
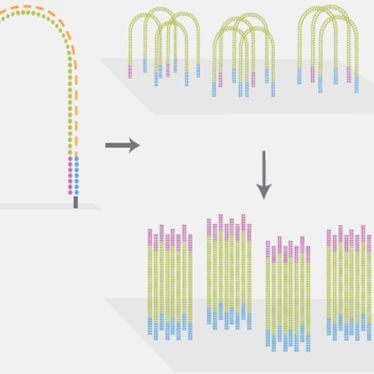
最近,一项最新研究成果为单细胞测序技术带来了突破,有望将单个细胞的测序成本降至 1 美分。SPLiT-seq 方法能兼容与甲醛固定的细胞或细胞核,且无需昂贵的专用设备,省去用定制化微流体或其他微孔分选单细胞的过程,而是用该细胞当作其自身 RNA 的隔离室。将细胞池分成多个组,引入成本低廉的组合条形码。SPLiT-seq 技术最关键的实现过程,其实就类似于一个 “洗牌” 的过程,通过这四次洗牌步骤,理论上可以得到 21,233,664 种条形码组合(在 96 孔板中进行三轮条形码编码,然后进行第四轮 PCR 反应,可以特异性的标记超过 100 万个细胞),最终给每个细胞一个独特的组合条形码 [33]。
SPLiT-seq 技术原理
技术难点及需要解决的问题
单细胞的获取:到目前为止,绝大多数单细胞转录组研究时还是需要单细胞悬液(比如组织解离液或者细胞培养悬液)做检测样品,细胞在解离、分选过程中,其转录状态是否发生改变,是要特别注意的一个问题。随之而来的一个问题是这种样品不能反映细胞在组织里的空间组织结构信息,除非我们知道这些细胞取自组织的哪一个部位。此外,各种各样的细胞构成了一个复杂的系统,进而形成了组织和器官的整体功能,解离状态的细胞打破这种整体性,因此我们还需要进一步发展微环境系统下单个细胞转录组的研究。
单细胞的 RNA 扩增:由于单个细胞里含有的 RNA 非常少,单细胞转录组研究工作主要依赖逆转录反应和扩增,这些反应非常容易出错或者丢失信息,以致产生了大量的偏差。大部分科研人员都会尽量减少 PCR 的反应循环数,就是为了减少这方面造成的误差,此外线性扩增技术还是能够在一定程度上解决这种因为扩增而带来的误差问题。
各种扩增技术带来的另一个不同是信息的保留,比如可以全长对转录本进行扩增和测序,这样能够整个基因及其不同转录异构体(transcript isoform)的结构信息,而我们也可以采取只对转录本的 5' 或 3' 端进行扩增的策略,只保留基因表达丰度的信息。再比如大部分扩增和建库过程都会丢失转录的方向信息,而这对基因表达定量的准确性,反义转录本以及其他非编码 RNA 的研究来说至关重要。最后,非编码 RNA 信息的保留也可能成为今后扩增方法改进的目标。
展望
近几年利用单细胞测序技术,在肿瘤异质性、免疫微环境、神经科学、胚胎发育、细胞分化等研究得到了优异的成果。但是,单细胞测序相关研究中还处在非常初级的阶段,同时也面临很多挑战。技术的不断改进和突变,将使单细胞测序技术在未来疾病精准研究与治疗具有十分广阔的应用前景。
伯豪生物经过 6 年的积累,建立了包括 SMART-seq,10X Genomics 和 BD Rhapsody 的单细胞 RNA 测序平台,为客户提供从样本保存、运输、单细胞悬液制备,到单细胞分选、建库和数据分析的一站式解决方案。目前已为来源于人、小鼠、大鼠、果蝇、斑马鱼、牛、羊等物种的临床和科研样本提供了单细胞测序和数据分析服务,研究成果发表在 Cell Research、Nature Neuroscience 等杂志上。伯豪生物将不断完善技术方法和服务体系,助力客户发表高水平研究论文。
伯豪单细胞测序数据分析展示
消除批次间差异
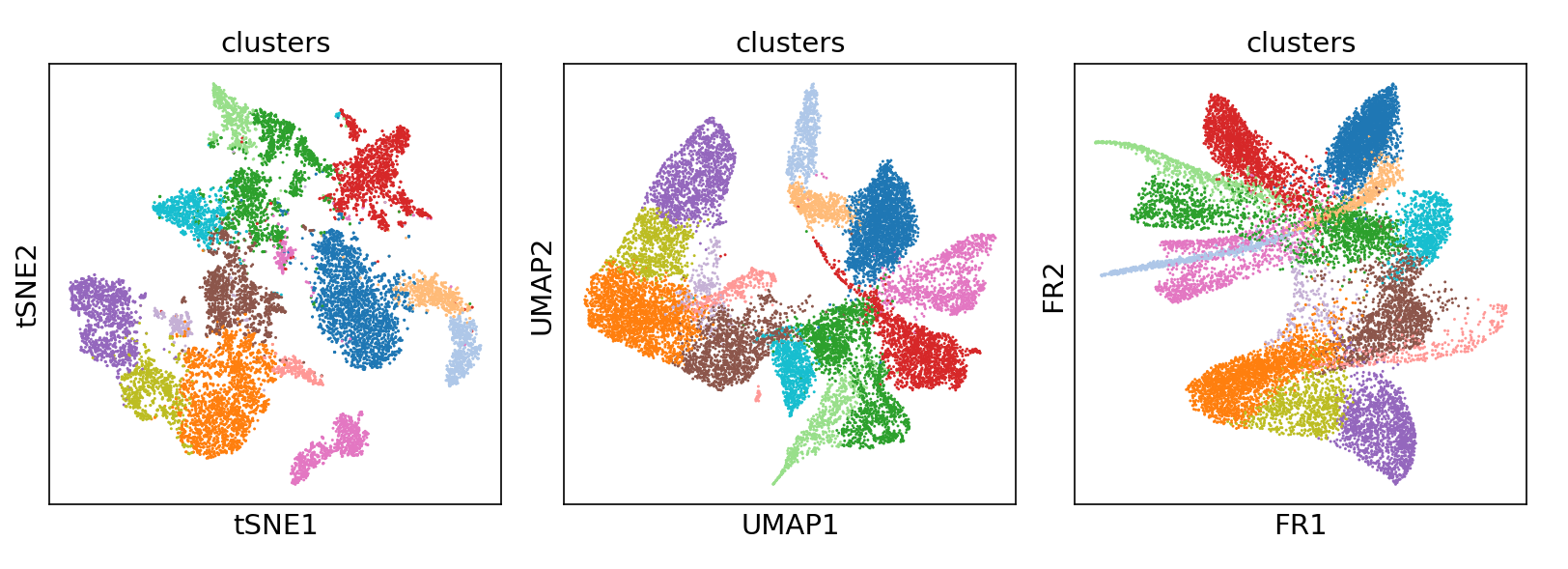
细胞亚群聚类及可视化
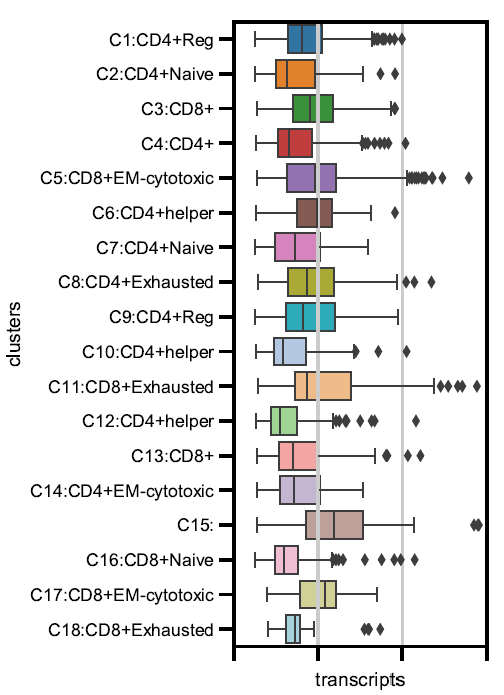
细胞亚群注释
不同 cluster 转录本表达丰度分布统计 不同 cluster 细胞数目统计
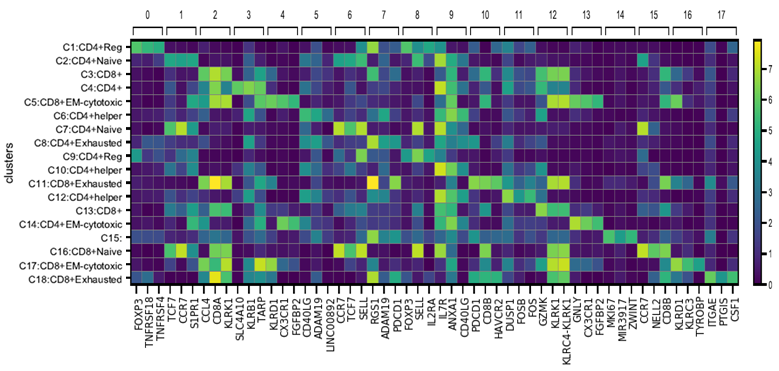
Marker 基因分析
拟时序分化转录因子调控网络
拟时序 GO 功能注释
患者之间的代谢特征差异分析
拟时序分析
参考文献[1] Tang F, Barbacioru C, Wang Y, Nordman E, Lee C, et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat Methods 2009, 6(5):377-382.
[2] Deng Q, Ramskold D, Reinius B, Sandberg R. Single-cell RNA-seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science, 343(6167):193-196.
[3] de Souza N. Single-cell genetics. Nat Methods 2013, 10(9):820.
[4] The biology of genomes. Single-cell sequencing tackles basic and biomedical questions. Science 2012, 336(6084):976-7.
[5] Method of the Year 2013. Nat Methods 2014, 11(1):1.
[6] Tang F, Lao K, Surani MA. Development and applications of single-cell transcriptome analysis. Nat Methods 2011, 8(4 Suppl):S6-11.
[7] Smith C. Cancer shows strength through diversity. Nature 2013, 499(7459):505-8.
[8] Harouaka R, Kang Z, Zheng SY, Cao L. Circulating tumor cells: advances in isolation and analysis, and challenges for clinical applications. Pharmacol Ther 2014, 141(2):209-21.
[9] Lovatt D, Ruble BK, Lee J, Dueck H, Kim TK, et al. Transcriptome in vivo analysis (TIVA) of spatially defined single cells in live tissue. Nat Methods 2014, 11(2):190-196.
[10] Sasagawa Y, Nikaido I, Hayashi T, Danno H, Uno KD, et al. Quartz-Seq: a highly reproducible and sensitive single-cell RNA sequencing method, reveals non-genetic gene-expression heterogeneity. Genome Biol 2013, 14(4):R31.
[11] Islam S1, Kjällquist U, Moliner A, Zajac P, Fan JB, et al. Characterization of the single-cell transcriptional landscape by highly multiplex RNA-seq. Genome Res 2011, 21(7):1160-7.
[12] Ramskold D, Luo S, Wang YC, Li R, Deng Q, et al. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat Biotechnol 2012, 30(8):777-782.
[13] Picelli S, Faridani OR, Björklund AK, Winberg G, Sagasser S, et al. Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc 2014, 9(1):171-81.
[14] Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344(6190):1396-401.
[15] Tirosh I, Izar B, Prakadan SM, Wadsworth MH, Treacy D, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352(6282):189-96.
[16] Grün D, Lyubimova A, Kester L, Wiebrands K, Basak O, et al. Single-cell messenger RNA sequencing reveals rare intestinal cell types. Nature 2015, 525(7568):251-5.
[17] Lee MC, Lopez-Diaz FJ, Khan SY, Tariq MA, Dayn Y, et al. Single-cell analyses of transcriptional heterogeneity during drug tolerance transition in cancer cells by RNA sequencing. Proc Natl Acad Sci U S A 2014, 111(44):E4726-35.
[18 Kim KT, Lee HW, Lee HO, Kim SC, Seo YJ, et al. Single-cell mRNA sequencing identifies subclonal heterogeneity in anti-cancer drug responses of lung adenocarcinoma cells. Genome Biol 2015, 16:127.
[19] Miyamoto DT, Zheng Y, Wittner BS, Lee RJ, Zhu H, et al. RNA-Seq of single prostate CTCs implicates noncanonical Wnt signaling in antiandrogen resistance. Science 2015, 349(6254):1351-6.
[20] Zheng C, Zheng L, Yoo JK, et al. Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell 2017, 169(7):1342-1356.
[21] Guo X, Zhang Y, Zheng L, et al. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat Med 2018, 24(7):978-985.
[22] Li L, Dong J, Yan L, et al. Single-Cell RNA-Seq Analysis Maps Development of Human Germline Cells and Gonadal Niche Interactions. Cell Stem Cell 2017, 20(6):858-873.
[23] Li CL, Li KC, Wu D, et al. Somatosensory neuron types identified by high-coverage single-cell RNA-sequencing and functional heterogeneity. Cell Res 2015, 26(1):83-102.
[24] Macaulay IC, Haerty W, Kumar P, et al. G&T-seq: parallel sequencing of single-cell genomes andtranscriptomes. Nature methods 2015, 12(6): 519-522.
[25] Angermueller C, Clark SJ, Lee HJ, Macaulay IC, Teng MJ, et al. Reik Parallel single-cell sequencing links transcriptional and epigenetic heterogeneity. Nat Methods 2016, 13(3):229-32.
[26] Hu Y, Huang K, An Q, Du G, Hu G, et al. Simultaneous profiling of transcriptome and DNA methylome from a single cell. Genome Biol 2016, 17:88.
[27] Hou Y, Guo H, Cao C, Li X, Hu B, et al. Single-cell triple omics sequencing reveals genetic, epigenetic, and transcriptomic heterogeneity in hepatocellular carcinomas. Cell Res 2016, 26(3):304-19.
[28] Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015, 161(5):1202-14.
[29] Klein AM, Mazutis L, Akartuna I, Tallapragada N, Veres A, et al. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 2015, 161(5):1187-201.
[30] M Soumillon, D Cacchiarelli, S Semrau, et al. Characterization of directed differentiation by high-throughput single-cell RNA-Seq. Anatomical Record 2014 , 217(1):16
[31] Fan HC1, Fu GK1, Fodor SP2. Expression profiling. Combinatorial labeling of single cells for gene expression cytometry. Science 2015, 347(6222):1258367.
[32] Zheng GX, Terry JM, Belgrader P, Ryvkin P, Bent ZW, et al. Montesclaros Massively parallel digital transcriptional profiling of single cells. Nat Commun 2017, 8:14049.
[33] Rosenberg AB, Roco CM, Muscat RA,Kuchina A, Sample P, et al. Single-cell profiling of the developing mouse brainand spinal cord with split-pool barcoding. Science 2018, 360(6385):176-182.







评论