产品详情
文献和实验
相关推荐
服务名称 :简化基因组测序2b-RAD
提供商 :上海欧易生物医学科技有限公司
2b-RAD技术是简化基因组测序技术的一次重大创新!
该技术由中国海洋大学王师教授于2012年在国际方法学期刊《Nature Methods》(IF: 25.328)上发表
一经发表即受到各领域学者极大关注。
截止到目前,利用该技术在国际国内学术期刊发表的文章已有一百多篇
其中不乏Science、MolecularEcology、Heredity、Proceedings of the Royal Society B等高水平刊物。
为使2b-RAD技术得到更好的推广应用
中国海洋大学海洋生物遗传学与育种教育部重点实验室将为青岛欧易提供长期的技术支持。
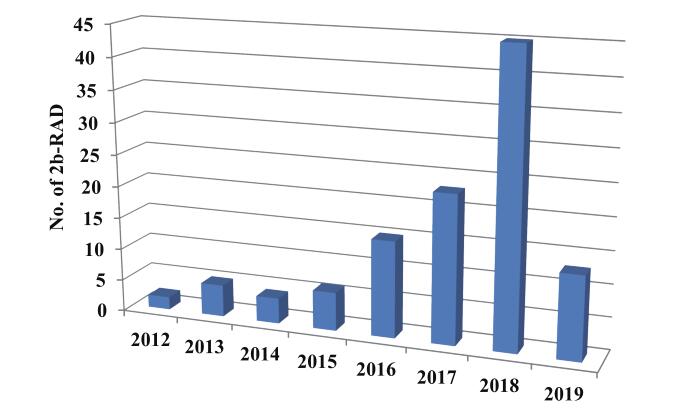
图1 2b-RAD技术发表的文章数量统计
该技术由中国海洋大学王师教授于2012年在国际方法学期刊《Nature Methods》(IF: 25.328)上发表
一经发表即受到各领域学者极大关注。
截止到目前,利用该技术在国际国内学术期刊发表的文章已有一百多篇
其中不乏Science、MolecularEcology、Heredity、Proceedings of the Royal Society B等高水平刊物。
为使2b-RAD技术得到更好的推广应用
中国海洋大学海洋生物遗传学与育种教育部重点实验室将为青岛欧易提供长期的技术支持。
图1 2b-RAD技术发表的文章数量统计
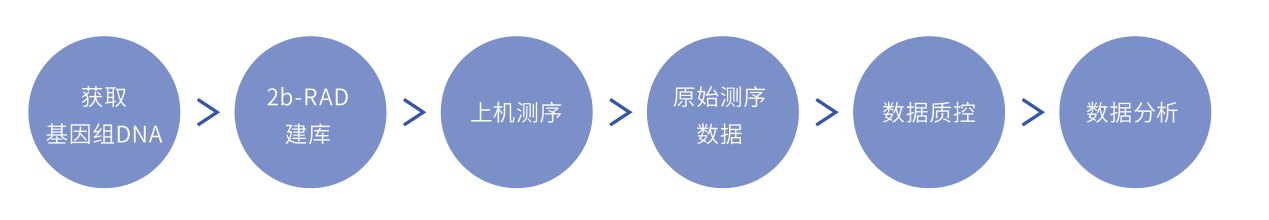
技术原理
2b-RAD技术使用IIB型限制性核酸内切酶(如BsaXI)对基因组DNA进行酶切,
切割位点位于识别位点的左右两侧,酶切后产生等长的33bp的标签,
这些标签经过富集后用于高通量测序,
通过生物信息学分析实现全基因组范围高通量SNP筛查和分型分析
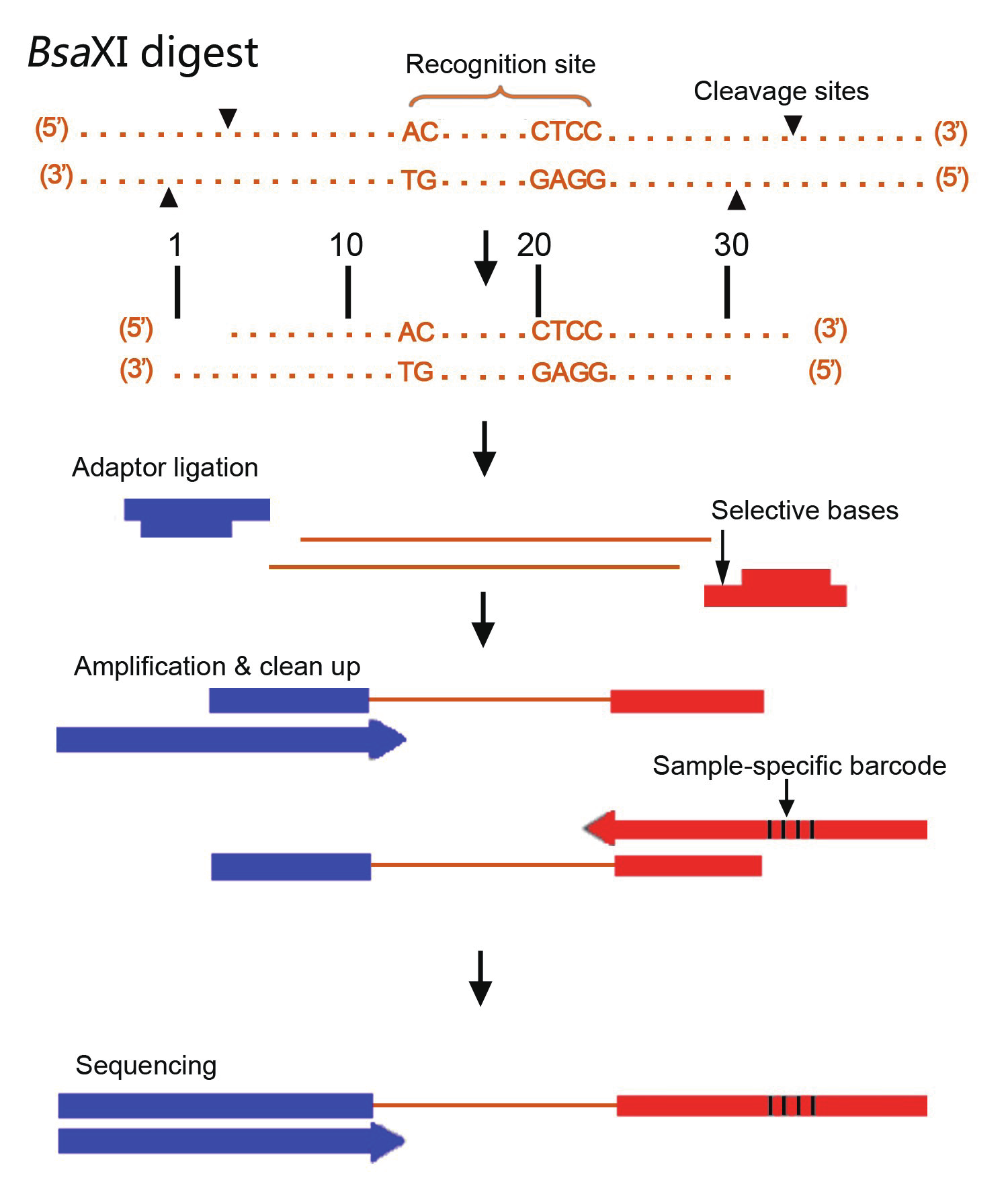
图2 2b-RAD技术流程
切割位点位于识别位点的左右两侧,酶切后产生等长的33bp的标签,
这些标签经过富集后用于高通量测序,
通过生物信息学分析实现全基因组范围高通量SNP筛查和分型分析
图2 2b-RAD技术流程
技术优势
所有酶切片段都用于测序
所有酶切片段都用于测序,避免了像其他RAD技术那样
必须经过片段选择而导致部分信息的损失
· 标签分布更均匀
无论是在拟南芥、人类基因组还是在扇贝基因组中
2b-RAD标签在除着丝粒位置外的整个基因组都有分布,并且分布非常均匀
· 测序深度均一
等长的33 bp酶切片段
PCR扩增效率和测序深度一致性好
保证了技术的高重复度

图3 2b-RAD标签在拟南芥、人和扇贝基因组上的分布及测序深度
· 标签密度灵活可控
一般平均3-4 kb一个33 bp的标签
可以通过选择不同的内切酶控制标签密度
两种及以上内切酶的组合可将标签密度增加到平均1 kb一个标签
也可以通过选择性接头(reduced tag representation ,RTR)
降低标签密度,增加测序深度
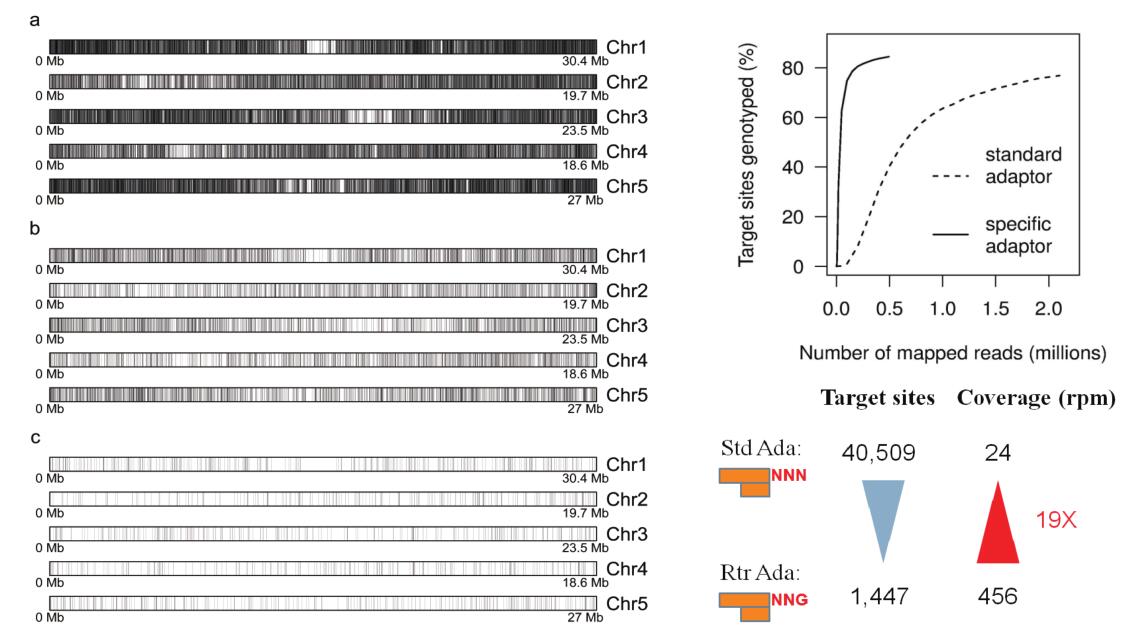
图4 不同内切酶和选择性接头两种方式调整标签密度
· 分型准确性高
对于无参考基因组的物种
基于混合泊松分布模型的全基因组de novo SNP 分型新算法(iML)
可有效去除重复序列对分型的干扰
与国际上主流算法相比分型准确率可提升高达20%
研发的RAD分型软件包(RADtyping),包含10余个软件组分
全面实现了从数据预处理至最终分型结果输出(显性和共显性标记)的全过程
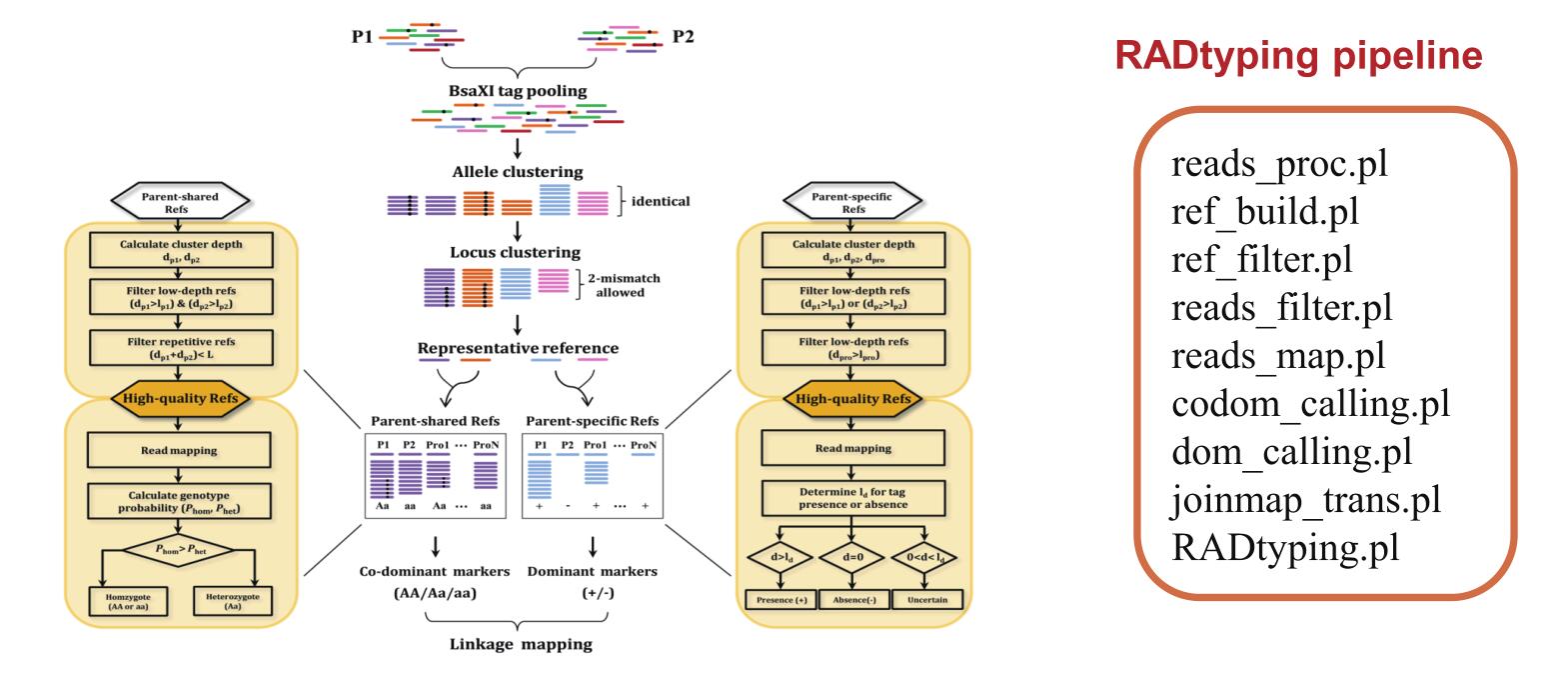
图5 基于RADtyping开发共显性和显性标记
· 样品质量要求低
降解样品、FFPE样品、标本等也可以检测。
青岛欧易已成功对中华绒螯蟹、虾等几个样品严重降解项目进行建库测序
并应用于遗传图谱构建、群体进化等研究

图6 2b-RAD技术成功应用于中华绒螯蟹(DNA严重降解)的研究
· 样品起始量低
建库起始量可低至1 ng

图7 不同DNA起始量的PCR扩增结果
· 助力发高水平文章
截止到2019年7月
2b-RAD技术已经发表了109篇文章
平均影响因子4.380
其中,五分之一的文章影响因子大于5.0
近一半的文章影响因子大于3.0

图8 2b-RAD技术部分已发表文章
必须经过片段选择而导致部分信息的损失
· 标签分布更均匀
无论是在拟南芥、人类基因组还是在扇贝基因组中
2b-RAD标签在除着丝粒位置外的整个基因组都有分布,并且分布非常均匀
· 测序深度均一
等长的33 bp酶切片段
PCR扩增效率和测序深度一致性好
保证了技术的高重复度
图3 2b-RAD标签在拟南芥、人和扇贝基因组上的分布及测序深度
· 标签密度灵活可控
一般平均3-4 kb一个33 bp的标签
可以通过选择不同的内切酶控制标签密度
两种及以上内切酶的组合可将标签密度增加到平均1 kb一个标签
也可以通过选择性接头(reduced tag representation ,RTR)
降低标签密度,增加测序深度
图4 不同内切酶和选择性接头两种方式调整标签密度
· 分型准确性高
对于无参考基因组的物种
基于混合泊松分布模型的全基因组de novo SNP 分型新算法(iML)
可有效去除重复序列对分型的干扰
与国际上主流算法相比分型准确率可提升高达20%
研发的RAD分型软件包(RADtyping),包含10余个软件组分
全面实现了从数据预处理至最终分型结果输出(显性和共显性标记)的全过程
图5 基于RADtyping开发共显性和显性标记
· 样品质量要求低
降解样品、FFPE样品、标本等也可以检测。
青岛欧易已成功对中华绒螯蟹、虾等几个样品严重降解项目进行建库测序
并应用于遗传图谱构建、群体进化等研究
图6 2b-RAD技术成功应用于中华绒螯蟹(DNA严重降解)的研究
· 样品起始量低
建库起始量可低至1 ng
图7 不同DNA起始量的PCR扩增结果
· 助力发高水平文章
截止到2019年7月
2b-RAD技术已经发表了109篇文章
平均影响因子4.380
其中,五分之一的文章影响因子大于5.0
近一半的文章影响因子大于3.0
图8 2b-RAD技术部分已发表文章
技术比较

项目流程
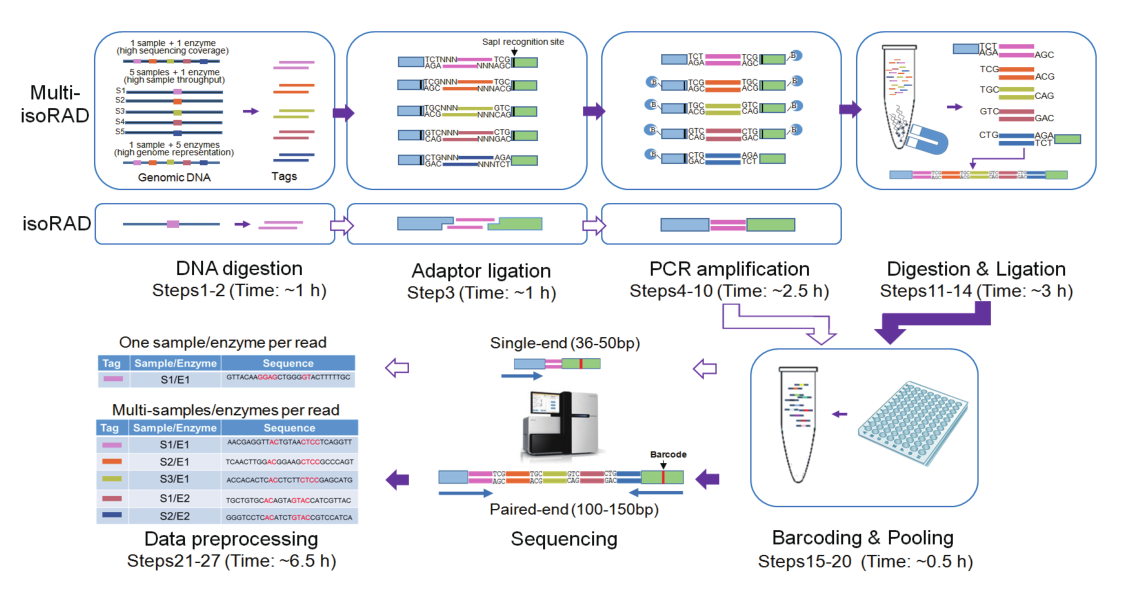
建库流程
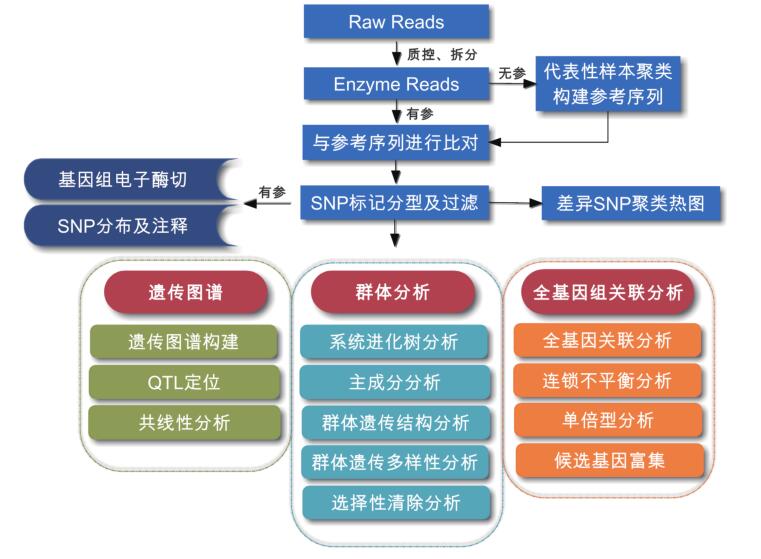
分析流程
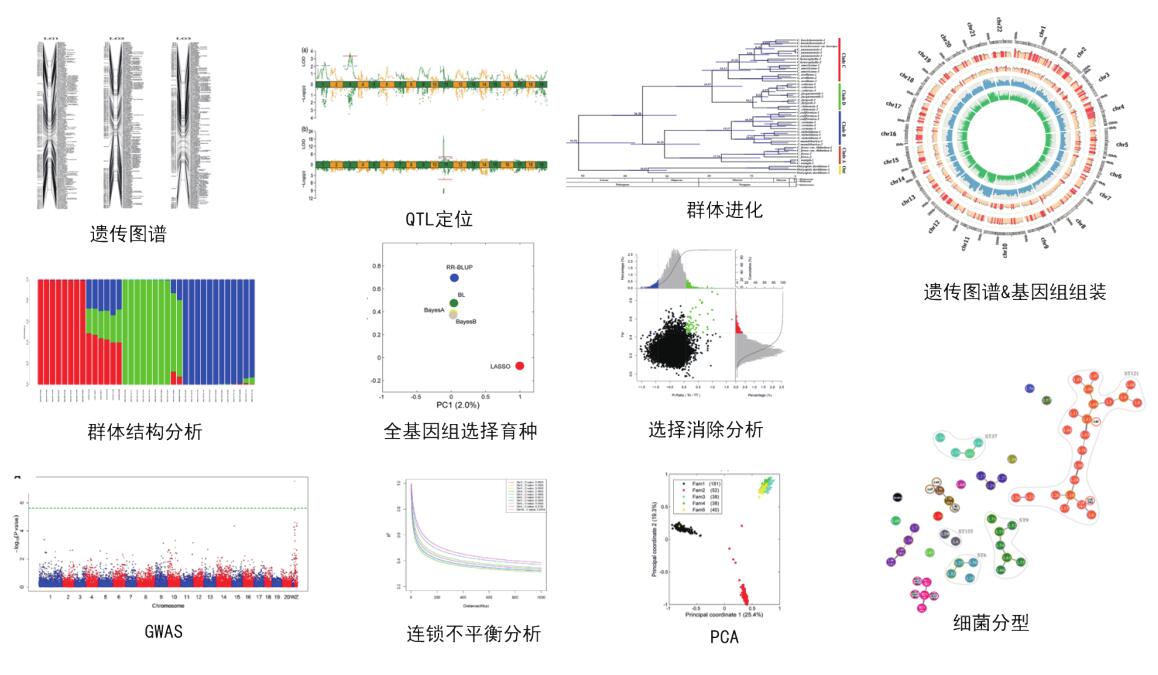
分析展示
常见问题
1. 2b-RAD中所用的酶是一种什么类型的酶?这种酶有什么特性?如何进行选择?
2b-RAD所使用的是IIB型限制性核酸内切酶,是一类商业化的酶。这类酶在基因组上识别特定的碱基序列(一般为5-7碱基),然后在识别位点两侧进行双链酶切,全基因组范围内切出的标签片段等长,如:内切酶BsaXI切出的为等长的33 bp片段。针对有参物种,可通过对基因组进行电子酶切结合预期的标签数确定合适的酶,对于无参物种则可选用其近缘物种基因组序列进行电子酶切。
2. 2b-RAD片段短,怎么获取标记所在位置的基因信息?
首先,不管哪一种简化基因组技术(RAD、ddRAD、2b-RAD),都存在片段短不方便设计引物的问题。但这个问题也不是没办法解决:1、比对到近缘物种基因组;2、进行基因组survey测序,然后比对;3、chromosome walking。随着测序价格越来越低,基因组survey测序不失为一种更好地方法。比如,Jiao et al. (DNA Research, 2013) 通过基因组survey覆盖栉孔扇贝全基因组的70%,将栉孔扇贝的转录组测序数据与基因组Scaffold进行了比对,88.4%转录本有功能注释结果,表明基因组预测序结果中已经包含了大部分基因。










上海欧易生物医学科技有限公司
品牌商实名认证
金牌会员
入驻年限:11年