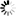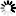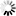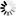线性化载体可以通过两种方法获得,不管采取哪种方法,最终线性化载体的 浓度需>15ng/μl。(高浓度的载体有利于提高效率)。
1、酶切选取合适的位点,单酶切或双酶切皆可,并且5´端突出,3´端突出或平末端均适用本Kit。
2、PCR 选取合适的位点,设计正向和反向引物,引物长度一般在18-20bp 左右,一般载体长度均大于3kb,建议用高保真的聚合酶扩增(推荐使用 Novoprotein pfu DNA 聚合酶,货号:E002)。为了避免模板质粒DNA对后续 试验的影响,建议载体酶切后再用PCR扩增,自连背景更低,阳性率更高。
目的片段通常通过PCR获得。引物设计要保证目的片段两端有至少15bp序列与 线性化载体的两端一致,特殊应用引物设计请参照技术手册。为保证PCR扩 增的特异性及灵敏度,请尽可能选用pfu等高保真酶(推荐使用Novoprotein pfu DNA聚合酶,货号:E002)。PCR每条引物长度至少在35-40bp,包括5´ 端与载体同源的15bp以及目的片段特异性序列20-25bp,(注意事项:如果是 表达载体克隆构建,引物设计完全后,请注意检查读码框的正确)。引物设 计方法:使用NovoRec®PCR一步定向克隆试剂盒(无缝克隆)时,引物设计是很重要的,在设计引物时同样要遵循引物设计的基本原则,只不过上下游引 物要加上15-18bp的载体同源序列。
载体同源序列如何添加主要分以下两种情况: (1)载体酶切后是5'端突出或平末端,则引物上的同源序列包括5'端突出部分的序列;(2)载体酶切后是3’端突出,则因无上的同源序列不包含突出部分。(如下图,红色碱基为希望保留原有的酶切位点所额外添加的碱基,不计算在15bp之内。
注:15bp同源序列不要求严格从载体最末端一届碱基计算,这是载体末端非同源序列将会被删除,同时导致重组效率的下降。当非同源序列达到20bp时,效率下降一半。

1.将目的DNA片段和线性化载体以一定的摩尔比加到管子中 进行重组反应
2.混匀后在37℃放置20分钟;
3.立即进行转化,剩余连接液可保存在4℃或-20℃待用。(注意:1.目的片段与载体的摩尔比在3:1-10:1之间,摩尔比低于3:1效率会降低 2.反应时间在20-40分钟,时间太长不利于重组反应)
建议所使用的感受态细胞效率要达到或>5X106 cfu/μg.
1.冰上融化一管100 μl的DH5a感受态细胞,轻弹管壁使细 胞重悬起来。加入10μl的反应液到感受态细胞中,轻弹数下,置冰上孵育45分钟。
2.42℃水浴中热激90秒后快速放入冰上5分钟。
3.加入500μl SOC液体培养基,37℃孵育45-60分钟。
4.5k离心3分钟收集菌体,根据需要将一定量的菌体均匀的涂布在含抗生素平板上。
一般的,我们建议采用菌落PCR进行阳性克隆鉴定(推荐使用Novoprotein 2x specificTM Master Mix 货号:E010)。
鉴定引物的选择:为避免假阳性结果,我们建议一条引物为载体特异性引物,另一条引物为目的片段特异性引物。