产品详情
文献和实验
相关推荐
提供商 :上海惠研生物科技有限公司
服务名称 : DNA甲基化芯片解决方案
规格 :欢迎致电询价。
高密度的升级版Human450K甲基化芯片实验服务与配套下游综合数据分析服务将围绕DNA甲基化组进行系统的生物学问题的探索。
表观遗传学标记(Epigeneticmarks)是由酶介导的DNA和相关染色质蛋白的生物化学修饰。尽管表观遗传标记不能改变DNA序列,但他们包含着遗传讯息并在调控基因组功能中发挥重要的作用。这类标记包括:胞核嘧啶甲基化(cytosinemethylation),组蛋白尾端和组蛋白核心的转录后修饰,以及核小体定(nucleosomespositions)。
他们影响着转录状态和染色质的其他重要功能,例如:DNA甲基化和某些组蛋白H3N端尾端的残基对转录基因的沉默和异染色质的形成起到关键性作用。这些标记针对具有细胞毒害作用的非编码基因序列的沉默,包括转座子(transposons),假基因(pseudogens),重复序列(repetitivesequences),以及整合的病毒而言就会被激活。表观遗传上的基因沉默在发育现象中,包括植物和动物中的印记基因现象,以及在细胞分化和重编程过程都中发挥重要的作用。
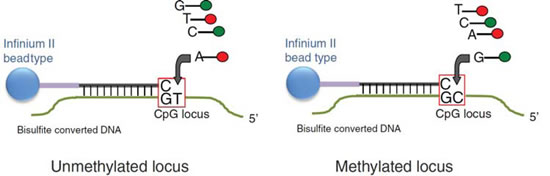
研究内容
DCAM-01:DNA甲基化芯片原始数据的预处理
DCAM-02:DNA甲基化芯片相关性与重现率(重复率/相关性)分析
DCAM-03:DNA甲基化芯片的处理组-对照组相关性评估
DCAM-04:样本组间的甲基化趋势差异分析
DCAM-05:样本组间的甲基化趋势(非监督/监督式)聚类分析
DCAM-06:基于甲基化谱构建疾病预后、诊断预测模型
DCAM-07:差异化甲基化位点临近基因生物通路分析
DCAM-08:差异化甲基化位点临近基因生物学功能注释
DCAM-09:DNA甲基化趋势与基因表达调控的关联分析
DCAM-10:DNA甲基化差异位点邻近启动子区调控基序(motif)的识别
DCAM-11:DNA甲基化趋势在TSS邻近区域的分布特征
DCAM-12:DNA甲基化趋势在基因组染色体上的分布特征
DCAM-13:MeDIP芯片技术与甲基化综合数据分析
DCAM-01:DNA甲基化芯片原始数据的预处理
根据Illumine Infinium methylation assay 甲基化芯片技术平台的特点, 针对每探针甲基化位点原始数据进行病例组和对照组样本组内的背景数值校正(background adjustment)和标准化(normalization)预处理,然后对探针水平以及基因水平指标分别进行均值计算。
我们采用Beta-value与M-value指标来表征Infinium methylation assay甲基化芯片中甲基化强度和水平。我们在后期差异甲基化分析是可采用基于Beta-value和 M-value的处理,一些研究表明Beta-Value衡量甲基化水平的方法在识别差异甲基化CpG位点(特别是高甲基化或非甲基化位点)时具有较大的异方差性。当降低最小差异阈值时,M-value方法比Beta-Value方法较好。我们在预处理时会多方面考虑如何选取甲基化强度和适当算法,以在后期计算差异甲基化位点时具有更稳健的结果。

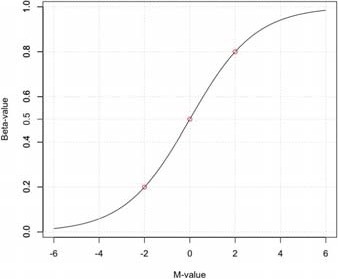 图-1M-Value与Beta-value关系曲线图
图-1M-Value与Beta-value关系曲线图

图-2实验样本的M-Value与Beta-value分布图
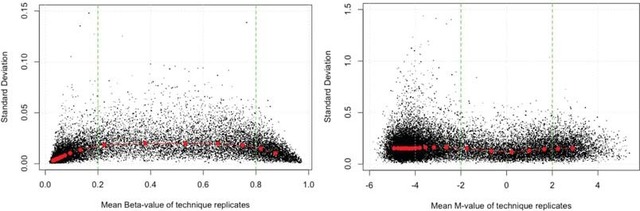
图-3实验样本的技术重复的均值M-Value与Beta-value与标准偏差分布图
DCAM-02:DNA甲基化芯片相关性与重现率(重复率/相关性)分析
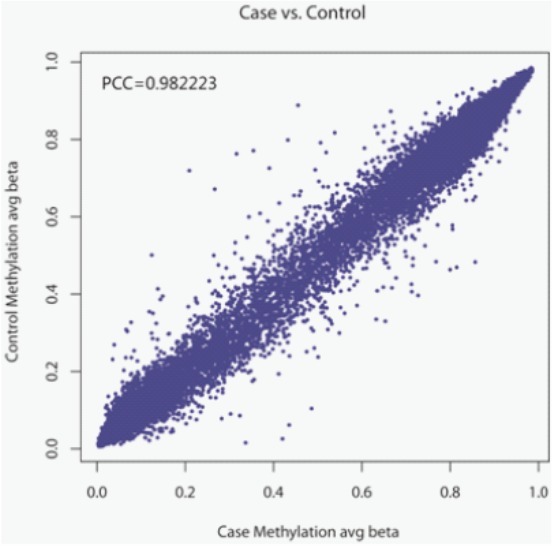 图 病例和对照组中甲基化强度整体的对比相关性计算分析
图 病例和对照组中甲基化强度整体的对比相关性计算分析
DCAM-03:DNA甲基化芯片相关性评估
依据各样本病例组和对照组的beta score,采用Fisher和Pearson卡方检验对病例组和对照组的甲基化位点和非甲基化位点分别进行统计,本分析期望探寻非甲甲基化与甲基化是否显著分布于特定的样本组内。
在基因水平上, 统计BeadChip芯片包含的甲基化位点(cpgID)和对应基因总数目。然后,计算每个基因在病例组和对照组中的平均beta score,再统计比对组中特有和共有的甲基化/非甲基化位点数目。我们采用Fisher和Pearson卡方检验对病例组和对照组的甲基化位点和非甲基化位点分别进行统计。
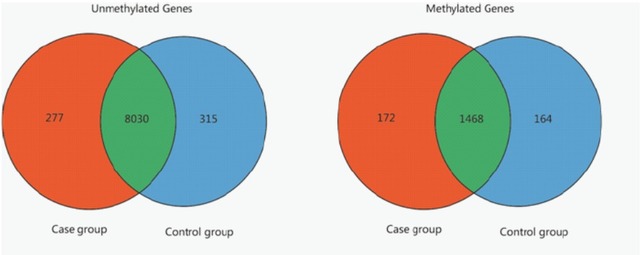
图甲基化/非甲基化基因在病例和对照组中数目比较
DCAM-04 :样本组间的甲基化趋势差异分析
实验组(或病例组)与对照组之间的差异化甲基化趋势分析将采用wilcox检验对每一甲基化位点的探针进行检验。对于多重假设检验的矫正,我们将分别采用多种校验算法,包括q-value, 配合beta-value, M -value的差值(实验组-对照组)通过排序和设定不同统计学显著性的筛选阈值的策略得到显著的待定差异化甲基化位点。原则上,我们选取p<=1E-2 作为差异甲基化位点的阈值,同时也会参考其他检验指标;原理是选择低p值保证高度可信的差异化位点。
另外,通过分位数-分位数图(简称Q-Q plot),我们也可以针对来自基因特异性甲基化模式-样本表型的关联分析得到的p-value(Observed -log(p-value))与原假设计算得到的关联性期望p-value(Expected -log(p-value))进行对比,从对比分布图上我们可直接得到与表型相关的特异甲基化基因的存在性。
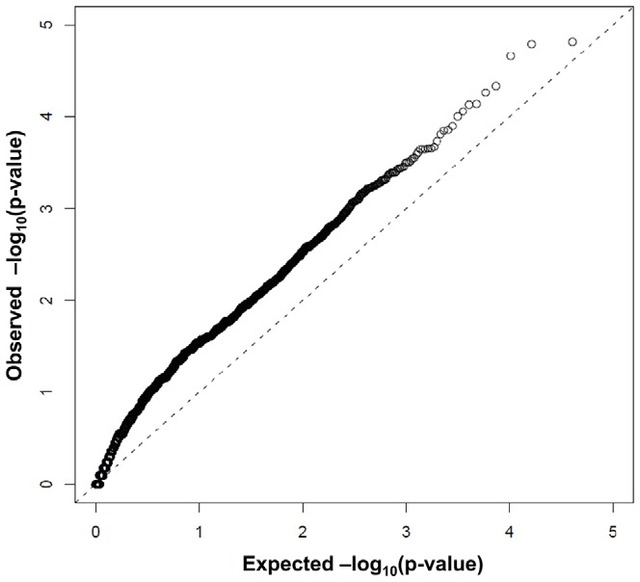
图-1 观测与期望p-value的分位数-分位数图(简称Q-Q plot)
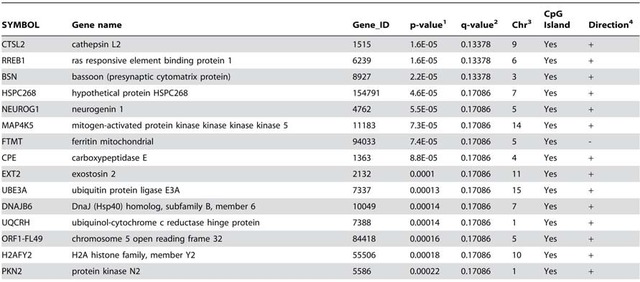
图-2 差异甲基化位点基因统计结果表
DCAM-05 :样本组间的甲基化趋势(非监督/监督式)聚类分析
本方案可对全基因组甲基化谱进行监督与非监督聚类分析,并可在聚类图下方用颜色标注出样本类型(注释。根据聚类的情况,行表示每个甲基化位点特征(即每行代表一个甲基化位点探针CpG ID),列表示样本。层次聚类选择对样本和位点同时聚类。我们发现聚类结果与构建基于SVM方法和甲基化指标的模型诊断预测性能相互吻合。
非监督聚类采用层次聚类( Hierarchical Manhattan distance和Average linkage方法);同时,本方案还可采用递归分区混合模型(RPMM-recursively partitioned mixture modeling)在所有样本中从基因组范围内对甲基化趋势进行分类判断(如下图所示)。通过重排检验(Permutation)方法进而判断特有的甲基化模式分类的统计学显著性p-value。
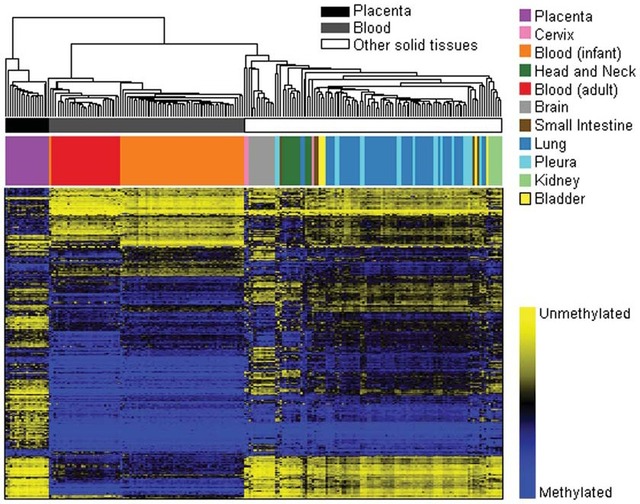 图-1采用层次聚类方法对样本甲基化趋势分类判断
图-1采用层次聚类方法对样本甲基化趋势分类判断
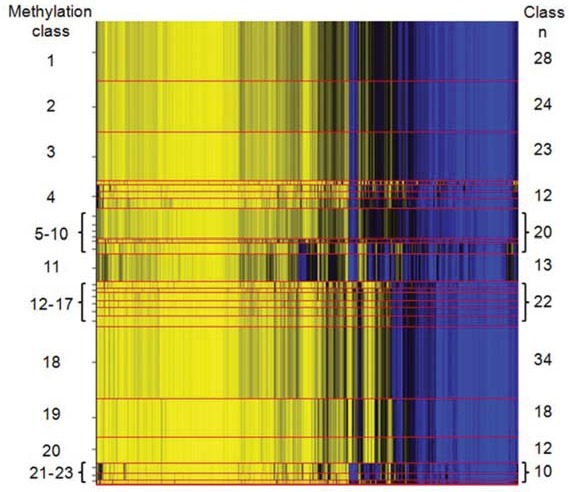
图-2采用递归分区混合模型(RPMM-recursivelypartitionedmixturemodeling)识别样本组
特有甲基化模式。行表示样本,样本分类结果采用红色线条分割开
本方案依据递归分区混合模型(RPMM-recursively partitioned mixture modeling)识别样本组特有甲基化模式。甲基化谱趋势类别(Methylation class#)与样本归属/共性之间的关联性,每样本在个类别所占比例等信息将被统计成表-1的结果形式。另外,我们也采用监督聚类方法,如随机森林(random forests)方法对样本群体的甲基化进行分类,并进行分类效果的统计(采用confusion matrix形式)。
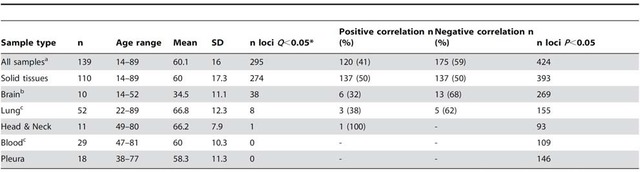
本方案可配合甲基化差异表达分析,也可以进一步地完成协变量(covariates),如年龄,生活历记录,生理或临床指标特征,与甲基化趋势之间的关联检验 (Association Test)。
表-1 采用递归分区混合模型(RPMM-recursively partitioned mixture modeling)分析
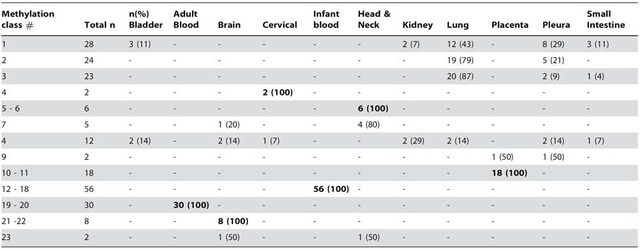
表-2 采用随机递森林(random forests)分类算法的样本分类统计
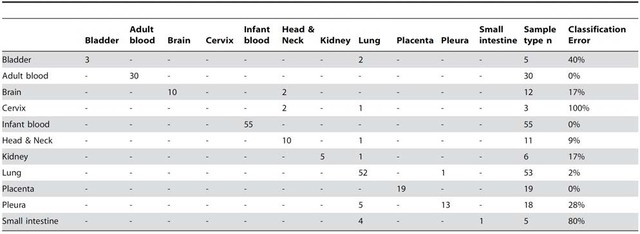
表-3 全局样本组特异的CpG甲基化模式与协变量(例如,年龄)关联分析统计结果
DCAM-06:基于甲基化谱构建疾病预后诊断预测模型
本方案可提供解决发现差异化DNA甲基化趋势的算法和计算分析技术,并依据差异化DNA甲基化谱构建疾病或表型的分类模型;特别的,对于病例对照设计实验的样本,我们可以构建用于临床预后或诊断的DNA甲基化分子预测模型。DNA甲基化的变异存在多种形式,各种形式的变异情况可能会导致疾病和其他个体表型的差异性。图-1给出目前通常被考虑到的DNA甲基化变异的类型。出现DNA甲基化变异的位点称为MVP: (Methylation variable position ),差异DNA甲基化区域称为DMR(Differentially methylated region),其中DMR可以发生于不同的情况下,例如:
• iDMR — imprinting-specific differentially methylated region
• tDMR — tissue-specific differentially methylated region
• rDMR — reprogramming-specific differentially methylated region
• cDMR — cancer-specific differentially methylated region
• aDMR — ageing-specific differentially methylated region.
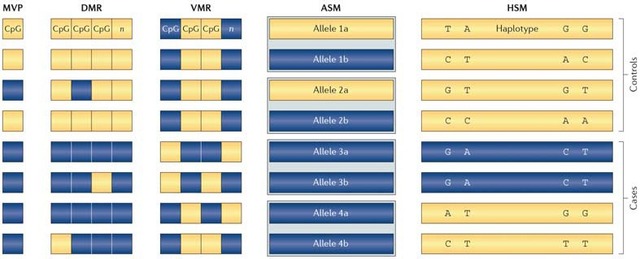
特异等位基因甲基化位点和区域存在甲基化变异称为ASM(Allele-specific methylation),主要来自父母DNA甲基化模式的变异,并与SNP多态性相关联。特异单倍型的甲基化变异(Haplotype-specific methylation ,HSM)则指具有共同遗传的SNP特征的差异化甲基化区域。另外,针对CpG岛(CGI)特征区域所定义的甲基化变异也将会被分析考虑;除了CGI区域,在CGI外沿的低密度CpG位点区域,称为CGI shore, 也可能出现较大的甲基化程度变异。综上所述,本方案将考虑多方面的甲基化变异情况采用SVM支持向量机理论中的多项式核算法技术加以识别和判断。
在本项目中我们采用简易和较为稳定的多项式分类器和SVM优化算法SMO[19-20]构建疾病特征预测模型。 由于统计学习理论和支持向量机建立了一套较好的有限样本下机器学习的理论框架和通用方法, 既有严格的理论基础, 又能较好地解决小样本、非线性、高维数和局部极小点等实际问题, 因此成为生物学领域的研究方向之一, 其核心思想就是学习机器要与有限的训练样本相适应,得到鲁能性的疾病诊断预测模型。
疾病预测模型分析结果与讨论
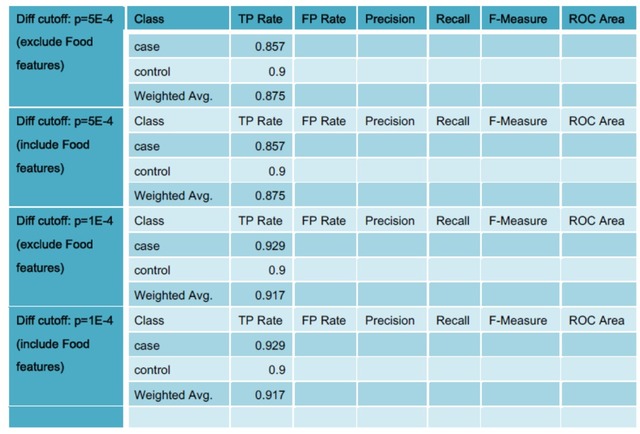
疾病模型的准确度评估(Summary Table-1)
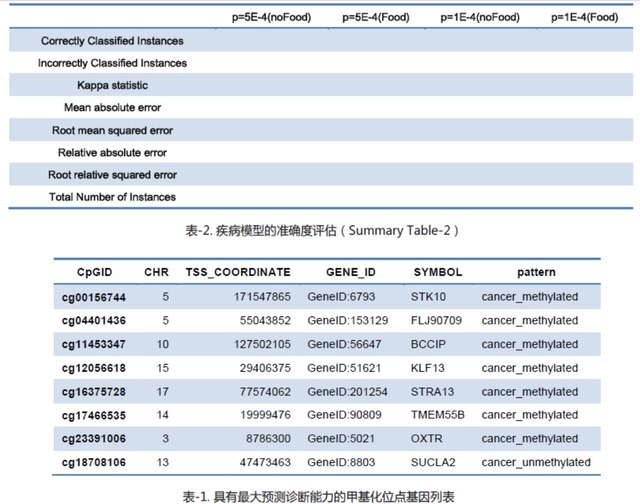
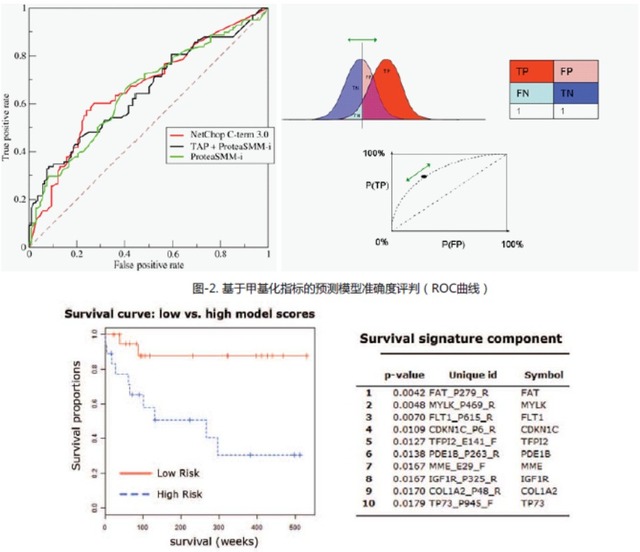
图-3.甲基化差异趋势结合生存随访数据进行生存分析
DCAM-07 :差异化甲基化位点临近基因生物通路分析
生物学通路(Biological pathway),包括代谢通路和信号转导通路是生物功能的重要组成部分,我们将各种形式的DNA甲基化基因的生物学通路进行综合分析,考察DNA甲基化变异对pathway的影响程度和影响的规律。通过GSEA(配合芯片表达谱数据),KS检验,超几何分布检验等方法对变异基因在某些pathway的富集程度进行排序,识别发生功能改变的潜在通路。生物学通路分析将采用以下pathway的资源,我们对所有信号通路进行整合优化,并适用于我们的个性化分析。
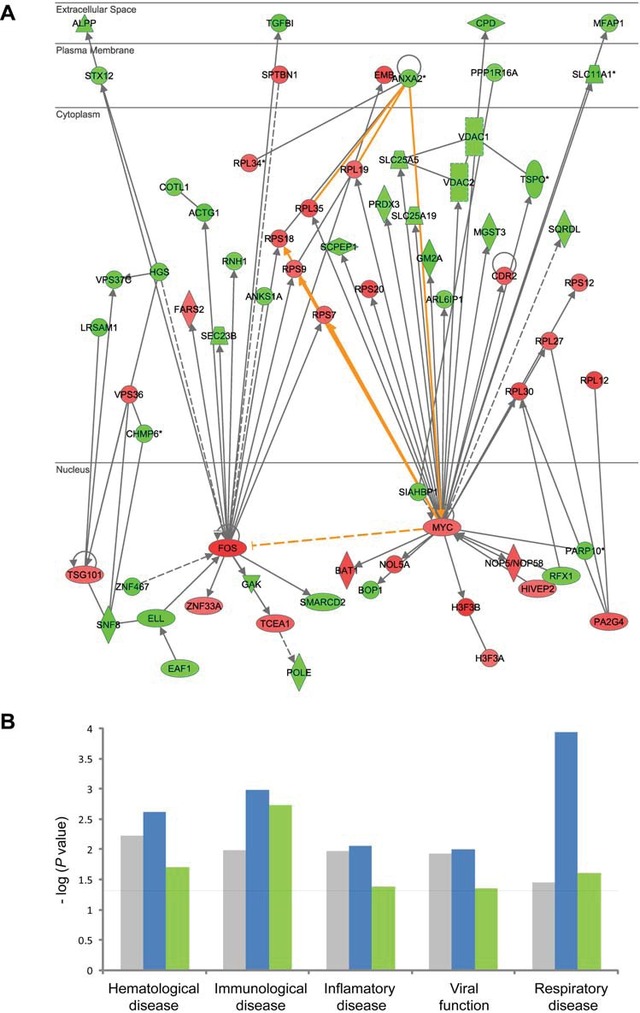
DCAM-08 :差异化甲基化位点临近基因生物学功能注释
本方案将对甲基化差异区域基因的功能进行注释分析,针对差异甲基化区域的分子功能生物学过程进行富集度分析(GO enrichment analysis)。根据超几何分布计算差异基因参与的生物学功能类别以及生物学通路。
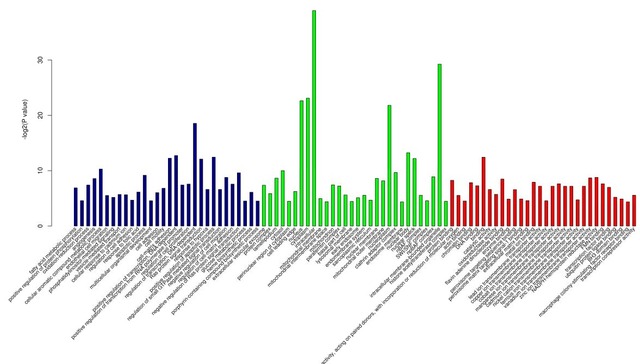
图例:GO 富集度分析(GO enrichment analysis)
DCAM-09 :DNA甲基化趋势与基因表达调控的关联分析
本方案针对基因转录水平和对应的基因甲基化程度进行关联分析,期望识别出甲基化空间/时间特异性与基因表达转录调控之间的联系。该分析模块需要结合基因转录组表达谱数据。可以通过RNA-Seq测序方法或表达谱芯片获取基因转录组表达谱数据。
在分析模块中,我们主要针对基因的上下游,基因体,外显子区的甲基化,内含子甲基化等不同分段序列进行甲基化基因比例的统计(图-A); 另外,针对假基因和非表达基因也进行甲基化程度和比例的计算(图-B)。结合基因的表达水平,我们对基因体内的甲基化,启动子区甲基化,非甲基化和全部情况分类进行讨论,计算各自情形在表达水平区间内的比例(图-C);最后,在样本对比的同时,利用信息熵值(Entropy)的概念计算样本(例如,组织tissue)特异性与基因体内甲基化,启动子区甲基化和非甲基化的各自情形比例(图-D)。类似的,我们针对基因的5’端边界和3‘端边界,分别对甲基化和非甲基化的启动子/基因区进行表达谱程度(纵轴)的刻画,分析甲基化位置特异性和强度与表达谱强度之间关联(图-E.F )
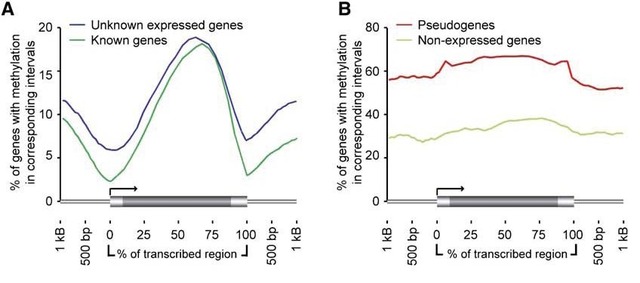
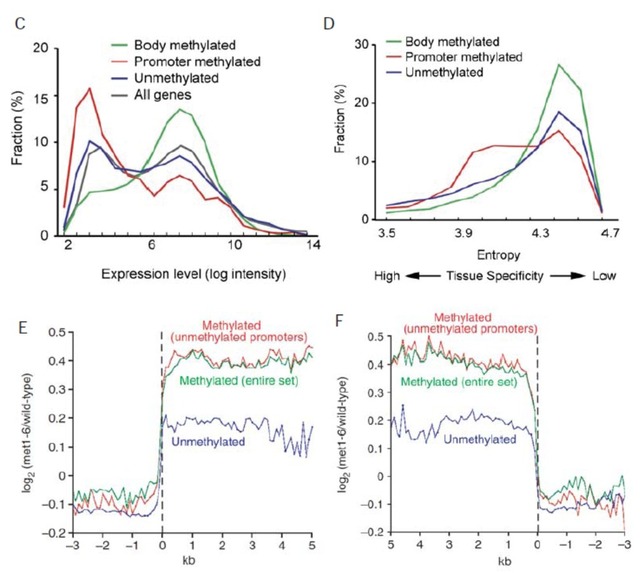
对甲基化差异区域基因表达情况和甲基化程度进行关联分析。我们还分析DNA甲基化与基因表达水平的关联性。对于实验组和对照组,我们将区分开来分别讨论关联性。同时,关联性分析只考虑场染色体内的具有显著DNA甲基化差异的CpG位点。
首先,对实际样本观测情况和随机模拟情况进行对比,分别统计各自密度函数,最后统计得到过甲基化基因中在转录水平上下调所占的比例(下图A,B)。另外,以CpG岛为考察对象,同样通过与随机情况对比识别在过甲基化CpG岛中具有下调基因所占的比例(下图C,D)。
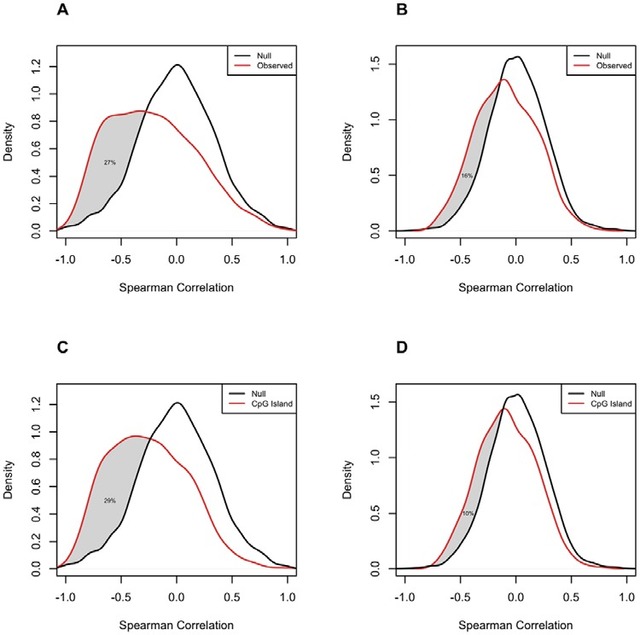
DCAM-10 :DNA甲基化差异位点邻近启动子区调控基序(motif)的识别
本方案可以识别出差异甲基化区域上存在的特殊DNA序列模式,DNA基序(motif)。这些序列特征可能与差异化甲基化的发生机制相关,也可能在表观水平改变后对转录调控起到调控作用。 系统全面地识别发现顺式转录调控元件(cis-regulatory element)是重构差异化甲基化基因的启动子区调控元件的重要途径。我们在该项目中将系统识别差异甲基化基因中潜在受到转录因子调控的结合位点区域。
首先,根据差异甲基化基因启动子序列中的序列模式特征进行提取和训练,获得完全基于序列预测的结合位点-Motif;然后,将预测的Motif与目前已知的转录因子结合位点数据(包括JASPAR版本和新的TRANSFACT)进行PWM位置得分矩阵比对,从而获得潜在调控差异甲基化基因的转录因子。
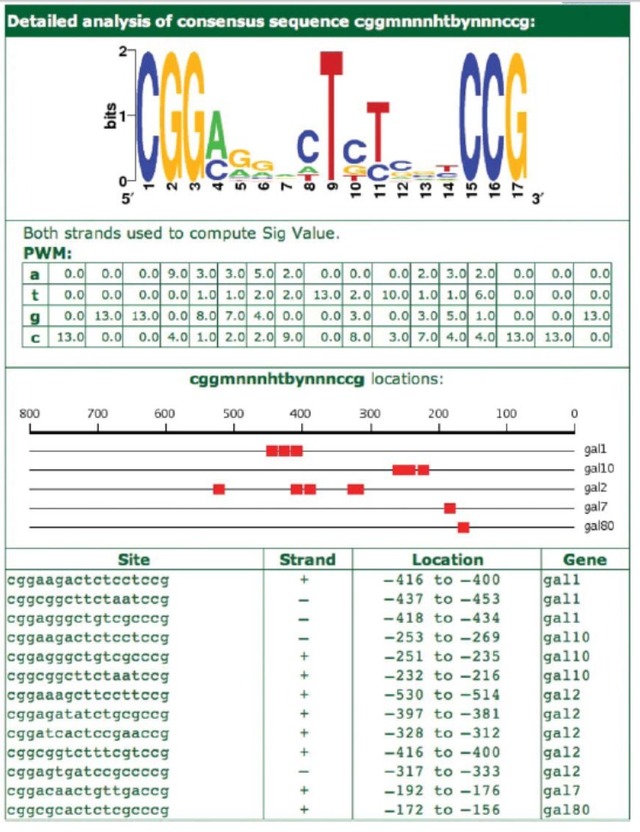
DCAM-11 :DNA甲基化趋势在TSS邻近区域的分布特征
本方案可对TSS临近启动子上下游区域上甲基化水平进行基因组范围内的统计分析,探讨甲基化调控在基因具体空间位置上的分布特征。结合基因表达水平数据,按照表达水平等级依次对甲基化的空间位置效应进行分类比较,方案注重细致地探讨基因组空间位置上的甲基化趋势。其次,也可以对基因组位点之间的甲基化相关性进行讨论,采用相关性分析技术,对不同距离内的CpG位点进行相关性分析作图。
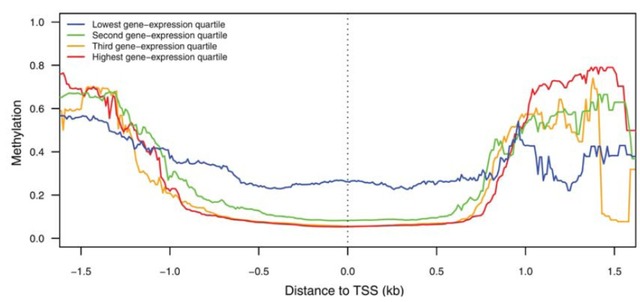
DCAM-12 :DNA甲基化趋势在基因组染色体上的分布特征
为了明确芯片探针的在UCSC hg19基因组版本中的定位,我们将芯片中的甲基化探针(长度各122bp)序列依次比对到人类基因组上,以确定唯一比配和多位置匹配的情况。其中唯一比对到基因组的甲基化探针具有高度特异性,其beta score信号值也是高度可信的;而多比对探针被过滤掉在后续分析中不予考虑(因为这种非特异性探针杂交后可能导致甲基化信号的假阳性结果产生,因此在后续讨论CpG的特性分析时不做考虑)。
根据human UCSC19基因组有关基因区域(Gene region),外显子-内含子(Exon/intron), 小RNA(miRNA region),转录起始位点(TSS region),重复片断(SD)以及结构变异区(SV)等各类型基因组区域的注释坐标信息, 将甲基化芯片经过比对所明确的唯一比对的高特异性探针(称为高可信度甲基化探针,high quality probe)进行不同基因组区域分布的数目统计(参见图1,2)
图-1. 450K甲基化芯片位点在基因组上的分布情况统计
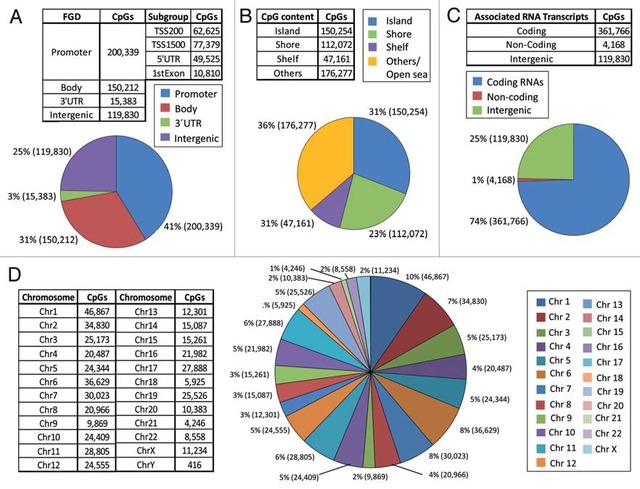
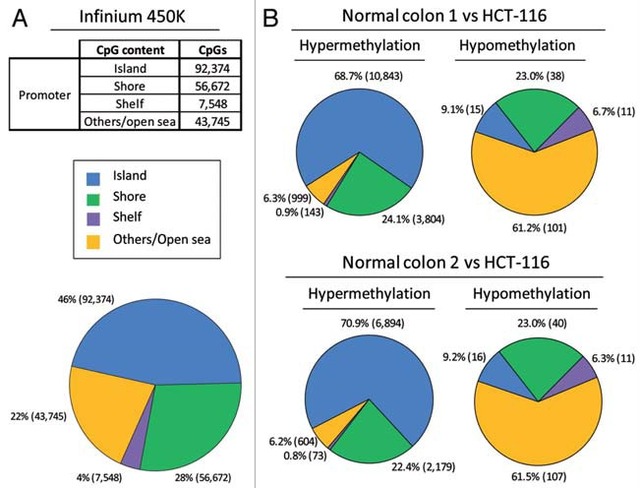
图-2 实验组与对照组在CpG岛空间位置效应下的甲基化水平对应比例分布统计
DCAM-13 :MeDIP芯片技术与甲基化综合数据分析
MeDIP(methylated DNA immunoprecipitation 甲基化DNA免疫共沉淀)技术已经被广泛应用于测定生物细胞在不同发育时期,临床表型,以及环境作用下的全基因组DNA甲基化状态研究。基于MeDIP的研究显示抗体富集度一定程度上反映决定了DNA甲基化水平,并且与RNA 聚合酶II的结合性(即转录活性的激活或者抑制状态)存在相关性。然而,通过MeDIP富集程度来准确的估计DNA甲基化水平仍然存在很多尚待解决的问题。
首先,MeDIP分析强制假设MeDIP富集度与DNA真实的甲基化水平呈线性相关,这种假设可能是错误的;其次,有研究表明DNA甲基化水平是抗体富集程度的函数,并且极大的取决于对应区域上的CpG整体含量。最后,MeDIP富集程度取决于富集片段与input片段之间的log比(即logR),然而这种度量却无法给出直观的解释,使得研究者武断的判断过甲基化或者低甲基化区域。因此,鉴于上述传统甲基化芯片分析的潜在问题,本方案将采用非线函数评估抗体富集度浓度(MeDIP enrichment)与真实的甲基化水平之间的关系,提高DNA甲基化水平预估数值的准确性。
本方案将对基因组上启动子类型根据CpG密度(CpG frequency)进行分类, 对不同启动子甲基化程度的各自特征进行分析, 包括
(1) High-CpG promoters (HCPs);
(2) low-CpG promoters (LCPs);
(3) Promoters with intermediate CpG content, 启动子具偏低水平CpG islands;
本方案适合于分析:NimbleGen 和Illumina,Affymetrix的DNA甲基化芯片
(1)芯片数据预处理: 芯片标准化处理: 组内标准化 (Loess based) 以及组间标准化处理 (Quantile based);
(2)芯片探针的基因组注释分析:每个探针的中心坐标将定位到基因组的已知的RefSeqs (来自UCSC human genome)上;7种不同的基因组区域将被考虑进入后续分析:包括: intergenic, intergenic,upstream,promoter, exon,intron, 5 UTR, 3 UTR.
(3)启动子注释分析
(a)对UCSC来源的启动子序列去冗余分析;包括去除长度延伸小于400bp的启动子,Y染色体上的启动子,以及缺乏足够证据支持的启动子,这些启动子会反映出基因间DNA甲基化的水平。
(b)筛选有效的启动子序列用于后续分析: 包括有 First Exon Finder (FirstEF) 启动子预测支持的证据,以及RefSeq记录支持的启动子;
(4) 启动子类别的定义与分类
HCPs (high-CpG promoters) : LCPs (low-CpG promoters); ICP( neither HCPs nor LCPs) (参见图例-2)
(5)分析三种启动子序列上甲基化程度与分布(参见图例-3),以及在不同样本之间三类启 动子的甲基化程度的分布(饼图)
(6)甲基化区域的基因功能分类分析
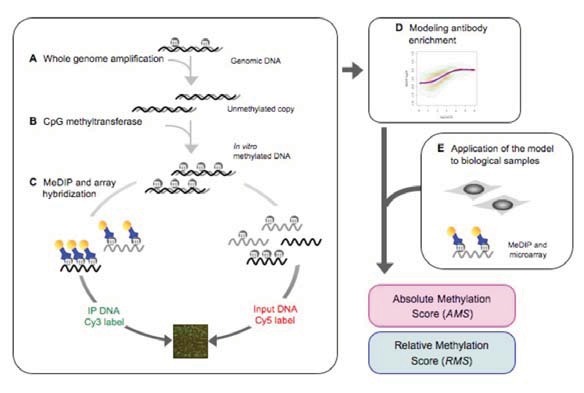
图例-1 (a) MeDIP实验策略数据分析流程
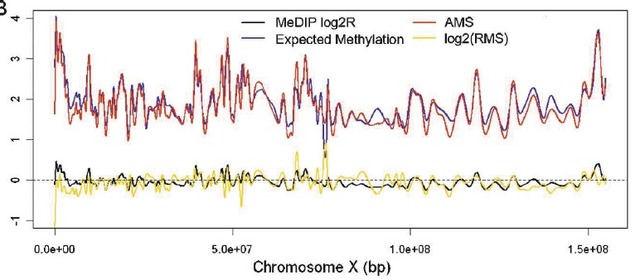
图例-1(b) 根据非线性模型预测的DNA甲基化水平峰值和在基因组上的坐标
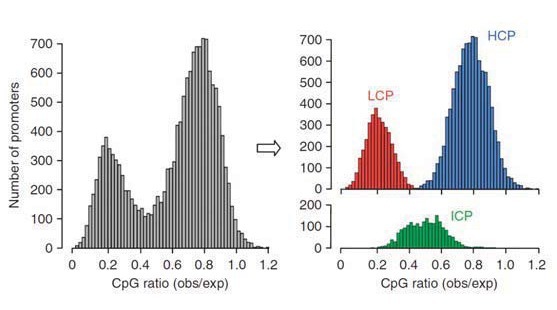
图例-2 全基因组启动子分类
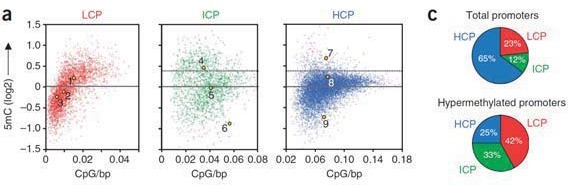
图例-3 不同启动子类别的甲基化水平及其分布
惠研云 | DaaS数据即服务 开创科学研究新思路
0 经验 0 背景的生物信息学小白也能立马上手
惠研生物致力于转化医学的技术研发服务为肿瘤基因组与疾病基因组提供强有力的大数据分析平台依据临床需求,为医生科室医院级别构建专病特色数据库通过AI遗传解读系统为临床患者提供自动化全方位解读病情,诊疗的方案,为患者获取更大获益的质量方案;为医生临床科研联动提供信息化平台。
其自主创新研发的“惠研云 BioGenius“平台,基于新一代测序技术,为科研工作者提供一站式生物医学大数据与临床转化服务:如实验方案(iProtocol), 实验服务,数据存储,医学统计与生物信息学自动计算分析平台,基础科研应用与转化医学解决方案,样本管理与项目管理等辅助信息系统等产品;满足从样本,实验,数据,分析到课题架构的全栈式开发服务,相应快速便捷的转化医学的研发需求。

我们的优势
-
拥有标准化操作实验室和高通量测序技术平台,实验周期短,质量可靠。
-
拥有Illumina HiSeq 2500、MiSeq等多种高通量测序平台。
-
技术人员经验丰富,可以根据合作伙伴要求提供实验方案、解决实验问题、分析实验结果。
-
拥有专业的生物信息团队和超算服务器,可为合作伙伴提供全面生物信息分析服务与技术支持。

惠研生物科技服务内容
-
科研方案咨询服务
-
课题基金申请指导
-
实验方案设计服务
-
整体实验实施服务
-
生物统计与数据挖掘
-
临床支持与转化服务
-
SCI论文发表全程支持
服务流程
整体实验技术服务分为两种类型:
-
当提供方案时,进行可行性评估并进行报价,然后进行反馈沟通,最终敲定方案执行;
-
当无方案时,根据研究热点和方向进行个性化设计,然后进行沟通从而确定方案。


为什么选择BioGeniusCloud进行生命科学和人工智能分析?
生命科学分析软件
丰富的数据科学工具库
为研究与论文发表提供高质量可靠保证
BioGenius 帮助科学家准确快捷的搜素感兴趣的生物分子基因和研究候选对象,加速了科学研究进程。我们了解高质量研究需要的软件和算法,可以提供可追溯,可重复验证和可发表的数据分析。因此,我们能为您提供可以信赖的数据科学工具和无代码化的部署和运用方式体验,帮助您更好的将精力用于课题和科学问题的设计和提出等更重要的方面。
惠研云 | 挖掘生命数字大数据产业价值
基于异构架构和超高速网络计算 | 成为科学研究第一推动力
BioGenius 惠研云平台,目前含有R语言包(>7万),Python包(>3万),Perl模块(>20万)等累计超过>500万个模块函数,满足需求、快速落地,程序自由搭建组合,并提供行业内细分领域丰富的最佳实践案例。惠研云平台已经对每个上线的程序完成了基准测试,可以按需提供智能连接和使用,实现线上快速部署,精准批量自动调度运行,以优化研发需求。借助惠研云AI智能分析工具,比如肿瘤基因组研究和新药研发等领域,可以实现在云端快速部署实验和运行分析,达到更快、更简单地完成研究任务 。
万物之始 大道至简 衍化至繁
惠研云 | 生物医学大数据计算平台 助力科研澎湃动力
惠研生物累计参与1000+科研项目
累计参与发表文献数目130多篇,累计影响因子919分
BioGeniusCloud 数据科学分析,帮助研究人员顺利登顶重磅学术期刊 发表高引用率论文获得同行认可。

研究领域
丰富的应用程序
通过惠研云无感知接口完成计算 | 从实验到数据得心应手
领先的数据驱动精准医学研发平台
DaaS数据即服务|提供科研澎湃动力
制定一份可靠的疾病研究路线图和目标识别战略耗时数月,在数据搜索、收集和解释等方面投入大量资源。告别传统繁琐模式,借助惠研云分析工具,可以更快,更简单地完成研究任务,这些工具可以在我们的云端快速部署和运行,探索数据的未知,并能够结合和利用科学界生成的所有科学数据解释 OMICs 数据。
同时,你无需花费时间试图将数据转换成可解读的格式或等待生物信息学家的解读,您可以通过惠研云平台访问和获取截至目前为止科学界生成的全部组学数据。所有的数据会以统一的方式进行处理和上传,让你能够完全专注于科学和数据解读。
数据为驱动|拓展可视化视野
想要获得科研的突破性优势,必须进行竞争性的实验设计以及周密严谨的分析,并可视化绘制出漂亮的报表与图。我们的分析报告就是这样做的。惠研云15年技术沉淀,利用知识驱动的数据库和无感知计算接口提供无偏差的图片和数据信息资源,用于推动科学研究的决策,制定研究策略,识别新的探索未知可能的机会和线索。
高性能节点服务器 | 分析无忧
基于Dell,超威等国际知名服务器供应商的支持,运用高性能刀片节点高密度计算性能,总计提供20000+核心算力,保证24x7连续生产力安全稳定。
Nvme SSD硬盘 | 让分析速度飞起来
基于Nvme SSD固态硬盘加速器,可以淋漓尽致发挥出网络计算的魅力,让你的任务计算飞起来!

高通量 | 高速率 | 无阻塞网络集群
高通量测序数据日益加大,呈指数型积累上升,必须交由高速网络提速分析。
基于以色列Infiniband技术,惠研云构建优化了集群计算性能,使得计算IO提升300%,持续数据交互速率提高到2T总带宽,保证集群中任务与数据0延迟,无阻塞。
深耕科研领域15年
国内数百家知名医疗机构信赖之选
惠研生物BioGenius为客户带来基因组的实时计算服务,构建全自动化云计算平台 。目前,惠研生物联合中国科学院生命科学院与全国三甲级医院科研团队共同研发 ,基于BioGenius 探索人类健康预防与机制的科学课题,人基因组学大数据, 多项成果陆续发表登录国际知名期刊,并与国内数百家知名企业深度合作,值得信赖!

点击上图了解“惠研云”生物医学数据计算平台
联系我们
上海惠研生物科技有限公司
官方网站:www.biogenius.cn
微信号:微信搜索“惠研生物”
客服电话::400-016-9606
技术咨询:180-1908-2932(微信同号)
企业邮箱:service@biogenius.cn








上海惠研生物科技有限公司
实名认证
金牌会员
入驻年限:10年